Kamagra gibt es auch als Kautabletten, die sich schneller auflösen als normale Pillen. Manche Patienten empfinden das als angenehmer. Wer sich informieren will, findet Hinweise unter kamagra kautabletten.
Ajur.uni.edu
AMERICAN JOURNAL OF UNDERGRADUATE RESEARCH
VOL. 11, NOS. 3 & 4 (2012-13)
SAR and Pharmacophore Based Designing of
Some Antimalarial and Antiretroviral Agents: An
INTERNET Based Drug Design Approach
Soumendranath Bhakat*
Department of Pharmaceutical Sciences
Birla Institute of Technology
Mesra, Ranchi-835215 INDIA
Received: April 26, 2012
Accepted: December 30. 2012
With the development of computational chemistry and molecular docking studies, Structure
Activity Relationship or SAR- and pharmacophore-based drug design have been modified to
target based drug discovery using sophisticated computational tools which is not very much user
friendly and has got many incompatibility issues with many operating systems (OS) and other
system configurations. In this paper SAR and pharmacophore based drug design approaches
have been described by the used of free internet based tools which are very much user friendly
and can almost compatible with any platform. Some antimalarial. And anti retroviral agents have
been designed using pharmacophore study and their drug like properties, toxicity, metabolic sites
and other parameters are predicted by the free internet based tools.
I.
rational drug design. These internet-based
tools are easy to handle because they use
In recent years computer-aided drug
JAVA to input structure and calculate the
[1-4] design has become an important tool
drug likeness and molecular properties in
for rational drug design on the basis of SAR
real time. These JAVA based internet tools
can be applied to predict the toxicity,
pharmacophore study. The SAR and
solubility, pKa, and Lipnski's five rule—all
pharmacophore-based drug design [5-6] is
important parameters for structure-based
mainly based on Lipski's rule of five.[7] But
rational drug design.
the high cost of the necessary computational
Recently structural analogue-based
software, its lengthy and complicated
drug discovery has become an important
installation, and system compatibility issues
tool for designing more potent drugs. This
all make SAR-proficient software difficult for
paper uses SAR, pharmacophore study, and
general undergraduate students to handle.
structural analogue-based novel drugs to
Most of these software packages are
design antimalarial anti-retroviral molecules
using internet-based tools.
WINDOWS versions (XP, 98, etc.) but are
MATERIALS AND METHODS
operating system, especially the 64 bit OS.
SAR-proficient software requires compilation
The structural analogue based drug
and a lengthy installation procedure, and
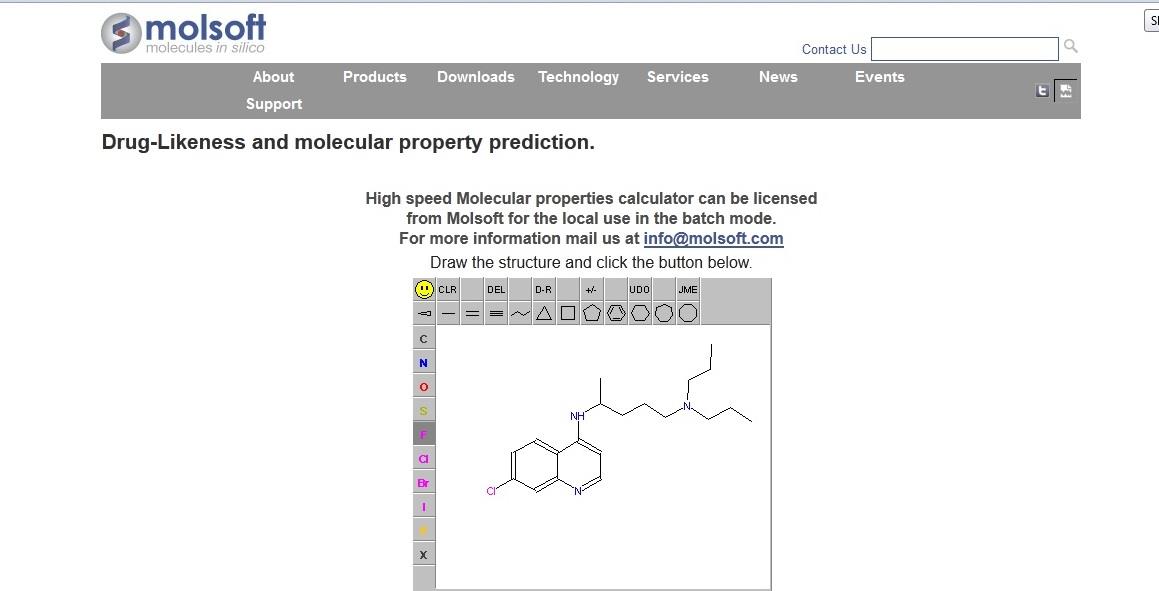
MOLSOFT, in which molecules'
in-silico
incompatibility issues with INTEL second
drug likenesses and molecular property
generation processors (i3, i5, i7, etc.).
prediction tool are important.[8] The new
To handle these difficulties in order
molecules, designed on the basis of SAR
to make drug design easier and convenient,
and pharmacophore study, were then
internet-based drug design on the basis of
inputted into a JME molecular editor [9] to
SAR and pharmacophores has become a
allow different properties to be calculated.
useful tool for modern structure-based
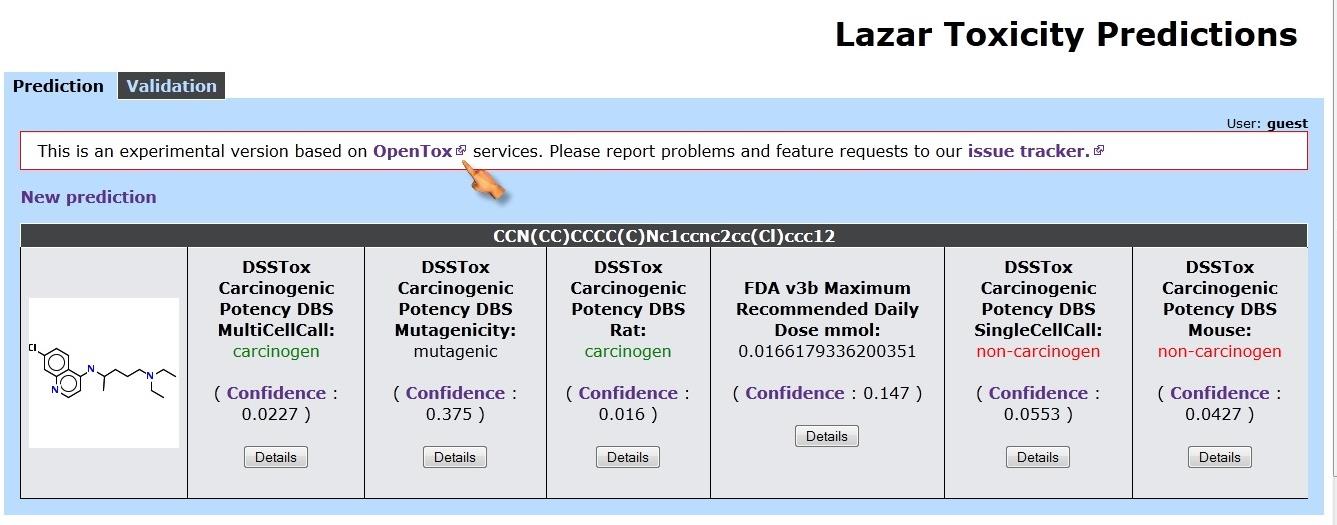
The lazar toxicity of all these designed drugs have been performed using
in-silico internet-
* Email:
[email protected]
based lazar toxicity prediction tools.[10] The
AMERICAN JOURNAL OF UNDERGRADUATE RESEARCH
VOL. 11, NOS. 3 & 4 (2012-13)
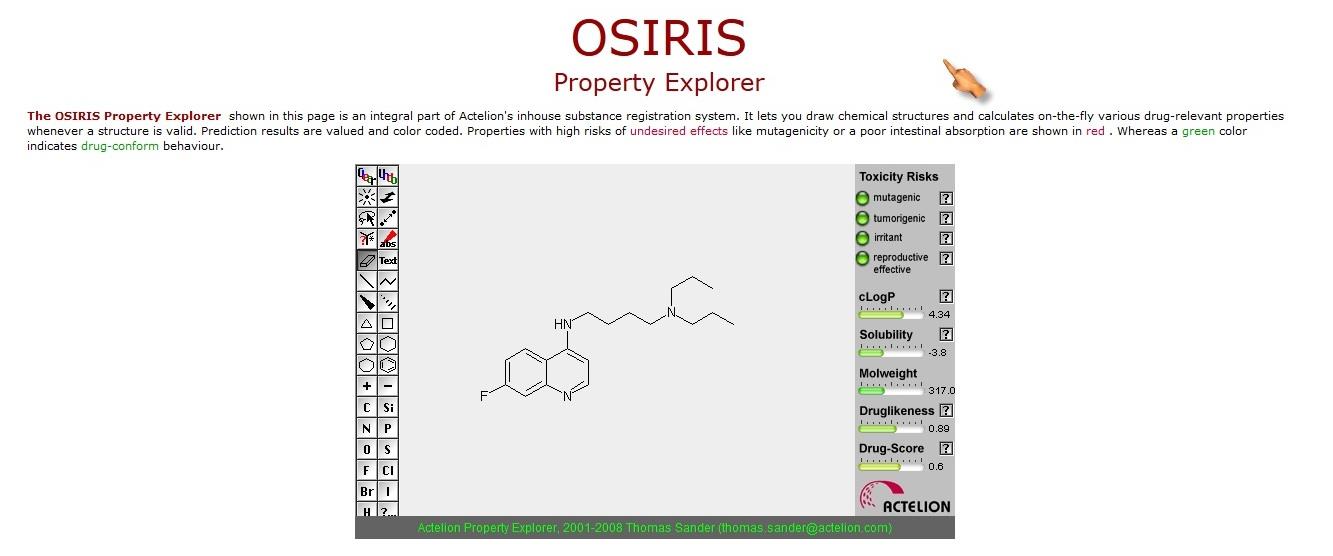
Figure 1. Screenshot of OSIRIS Property Explorer.
Figure 2. Screenshot of MolSoft Drug likeness and Molecular Property prediction tool.
Figure 3. Screenshot of Lazar toxicity prediction tool.
AMERICAN JOURNAL OF UNDERGRADUATE RESEARCH
VOL. 11, NOS. 3 & 4 (2012-13)
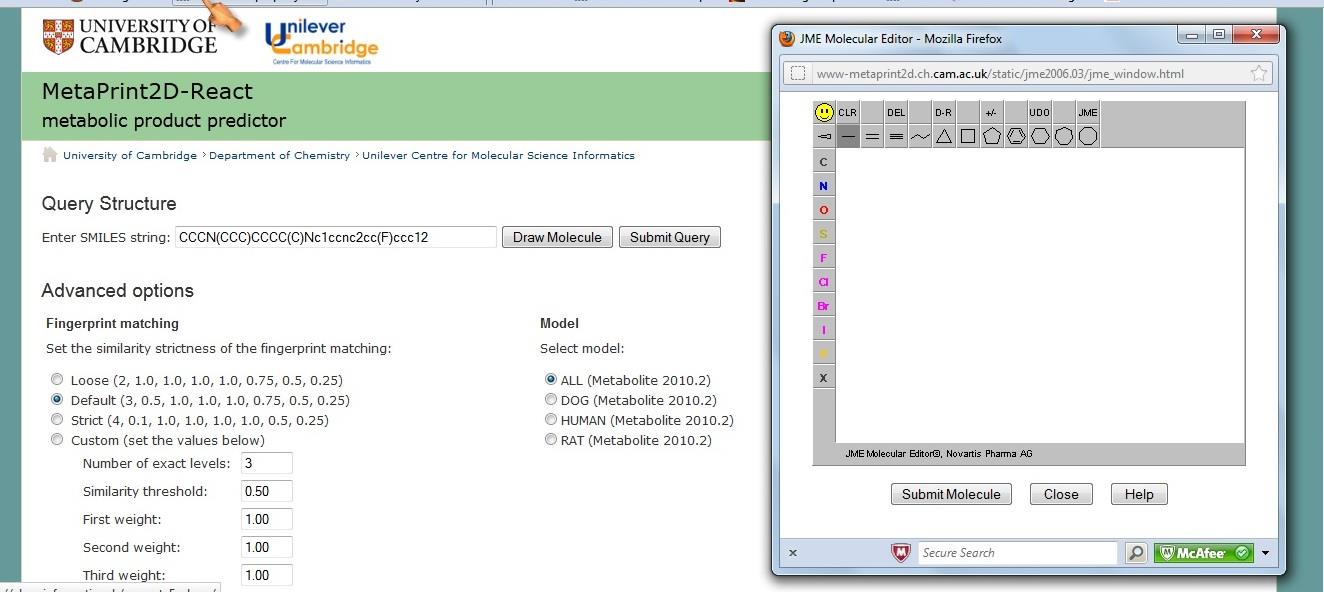
Figure 4. Screenshot of MetaPrint2D metabolic site prediction tool.
Figure 5. The 4-Amino quinolone pharmacophore, R1,R2 = positions of substitution.
OSIRIS Property Explorer [11] was also
kept malaria in check in most regions for
used to predict toxicity and other drug-like
decades. However, the rise in malaria
properties. The metabolic sites of these
deaths is due in part to the diseases'
designed drugs have been predicted using
increased resistance to chloroquine [14],
MetaPrint2D. [12] All these calculations
hence there is a necessity to design some
were performed using a WINDOWS 7 64-bit
more potent 4 aminoquinoline derivatives to
operating system having Intel Core 2 duo
introduce a more potent therapies.
The first type of drug designed using
internet based tools using chloroquine as
Results and Discussions
prototype drug having probable antimalarial
activity as it consists of 4-aminoquinoline
a. Designing of New Chloroquine structural
analogues as probable antimalarial
designed in such a way that it will show
Inexpensive and stable antimalarial
drugs such as the chloroquine [13] have
AMERICAN JOURNAL OF UNDERGRADUATE RESEARCH
VOL. 11, NOS. 3 & 4 (2012-13)
Molecular properties* of AM1: Molecular formula: C19 H28 Cl N3
Molecular weight: 333.20
Number of HBA: 2 Number of HBD: 1 MolLogP : 5.90 (> 5) MolLogS : -5.71 (in Log(moles/L)) 0.65 (in mg/L)
MolPSA : 24.58 A2
MolVol : 343.53 A3
Figure 6. Prototype molecule chloroquine
Number of stereo centers: 0
having MolSoft drug likeness score of 1.17
more drug likeness score than the prototype molecule
pharmacophore essential for the antimalarial
activity. The side chain present at 4 position of chloroquine have been modified with alteration of halogen atom in some cases at position 8 to get increased drug likeness
score with the MolSoft Drug Likeness and
Molecular Property prediction tool. In case of
Drug likeness* score 1.25
designed molecules AM3, AM4 the chlorine
molecule at position 8 have been replaced
Molecular properties* of AM2:
by –F atom to increase the MolSoft drug
Molecular formula: C20 H30 Cl N3
likeness score than the prototype molecule
Molecular weight: 347.21
Number of HBA: 2
chloroquine analogues with drug likeness
Number of HBD: 1
MolLogP : 5.35 (> 5)
molecular properties have been listed below.
MolLogS : -5.89 (in Log(moles/L)) 0.44 (in
The position of R1 and R2 in the 4-
aminoquinolone ring (Figure 5) are modified
MolPSA : 24.04 A2
in these designed molecules to get
MolVol : 359.14 A3
increased drug likeness score.
Number of stereo centers: 1
a. List of Designed Chloroquine Analogues
with Molsoft Drug Likeness Scores and Predicted Molecular Properties
properties, as predicted by MolSoft, are marked with an asterisk (*).
Drug likeness* score 1.23 Molecule ID:AM3
Molecular properties* of AM3: Molecular formula: C19 H28 F N3 Molecular weight: 317.23 Number of HBA: 2 Number of HBD: 1
MolLogP : 5.27 (> 5)
MolLogS : -5.13 (in Log(moles/L)) 2.37 (in
Drug likeness* score 1.38 Molecule ID: AM1
AMERICAN JOURNAL OF UNDERGRADUATE RESEARCH
VOL. 11, NOS. 3 & 4 (2012-13)
Molecule ID
cLogP value
Solubility
Molecular
Likeness
Table 1. OSIRIS Property explorer values of designed chloroquine analogues. All parameters in
OSIRIS property explorer shows "green" colour for all designed drugs which indicates the drug
confirm behavior of these designed chloroquine analogues.
MolPSA : 24.58 A2
(NOR). A high NOR indicates a more
MolVol : 332.26 A3
frequently reported site of metabolism in the
Number of stereo centers: 0
occurrence ratio does not indicate how likely
a molecule is to be metabolised, but rather the
occurring at a particular site in the molecule, assuming it is metabolised. The Indication
of colors which denotes predicted metabolic sites (see the Results Color Scheme on p. 39).
On the basis of predicted lazar
toxicity it is clear that the molecules AM3
Drug likeness score* 1.20
chloroquine,AM1 and AM2. But as the
Molecule ID: AM4
toxicity level of the drugs AM1 and AM2
Molecular Properties* of AM4:
have almost similar with chloroquine and in
Molecular formula: C20 H30 F N3
some cases the confidence value for the
Molecular weight: 331.24
predicted toxicity is more than that of
Number of HBA: 2
chloroquine (the confidence value for the
Number of HBD: 1
non-carcinogenic property of AM1 and AM2
in case of SingleCellCall** and Mouse**
MolLogS : -5.31 (in Log(moles/L)) 1.61 (in
parameters are more than that of the
chloroquine) hence they are believed to be
MolPSA : 24.04 A2
more potent than chloroquine as well.
MolVol : 347.87 A3
Number of stereo centers: 1
0.66 <= NOR <= 1.00
b. Phase I Metabolic site prediction using
MetaPrint2D by setting the strictness of
0.33 <= NOR < 0.66
the fingerprint matching in "DEFAULT"
and selecting model "ALL
0.15 <= NOR < 0.33
0.00 <= NOR < 0.15
The color highlighting an atom
indicates its normalized occurrence ratio
Results Color Scheme
AMERICAN JOURNAL OF UNDERGRADUATE RESEARCH
VOL. 11, NOS. 3 & 4 (2012-13)
Figure 7a. Predicted Phase I metabolic sites
Figure 7c. Predicted Phase I metabolic sites
Figure 7b. Predicted Phase I metabolic
Figure 7d. Predicted Phase I metabolic
AMERICAN JOURNAL OF UNDERGRADUATE RESEARCH
VOL. 11, NOS. 3 & 4 (2012-13)
c Potency
Potency DBS
Potency DBS
Recommended
Potency DBS
c Potency
Daily Dose
Table 2. Predicted toxicity of chloroquine analogues using in-silico lazar toxicity prediction tool
http://lazar.in-silico.de/predict.
IV.
DESIGNED ARTEMISININ
ANALOGUES AS ANTIMALARIAL
Artemisinin [16] (Fig. 8) is a drug
that posses the most rapid action of all
falciparum malaria. To increase the potency
(the main aim is to cure Artemisinin resistant
malaria [17]) and make this drug more broad
spectrum antimalarial agent the carbon at
position 10 of Artemisinin is replaced by
nitrogen with a side chain having a
analogous chloroquine like side chain and a
chlorine atom is attached with position 15 of
Artemisinin. The new designed drug like
molecules have been found to have more
drug likeness score than that of the prototype drug Artemisinin (Artemisinin has
a drug likeness score that of 1.22). The
toxicity and metabolic sites of these
designed drugs have been predicted using
the toxicity, metabolic site prediction tools.
Figure 8. The prototype drug Artemisinin.
AMERICAN JOURNAL OF UNDERGRADUATE RESEARCH
VOL. 11, NOS. 3 & 4 (2012-13)
a. List of Designed Artemisinin Analogues
with MolSoft Drug Likeness Scores and
MolPSA : 55.19 A2
Predicted Molecular Properties
MolVol : 443.65 A3
Number of stereo centers: 7
Molecule ID: AMM1
Drug-likeness score of AMM1: 1.97
Molecular Properties of AMM1:
Molecule ID:AMM3
Molecular formula: C
Drug likeness model score of AMM3:1.46
Molecular weight: 395.27
Molecular Properties of AMM3:
Number of HBA: 6
Molecular formula: C21 H37 Cl N2 O4
Number of HBD: 0
Molecular weight: 416.24
MolLogP : 5.46 (> 5)
Number of HBA: 6
MolLogS : -2.52 (in Log(moles/L)) 1202.47
Number of HBD: 0
MolPSA : 51.18 A2
MolLogS : -1.62 (in Log(moles/L)) 10018.30
MolVol : 445.00 A3
Number of stereo centers: 7
MolPSA : 42.77 A2
MolVol : 446.57 A3
Number of stereo centers: 8
Artemisinin and all its analogues
have shown mutagenic, tumorigenic and
irritant property as it shows in "red colour" in
OSIRIS Property Explorer but all of their designed analogues have more drug score
than that of Artemisinin hence it is predicted that these newly designed drug like
molecules have much more potency than
that of the prototype drug Artemisinin.
b. Phase I Metabolic site prediction using
MetaPrint2D by setting the strictness of the fingerprint matching in "DEFAULT"
Molecule ID: AMM2
and selecting model "ALL (Metabolite
Drug-likeness score of AMM2: 1.90
Molecular Properties of AMM2:
Molecular formula: C21 H36 N2 O5
The color highlighting an atom
Molecular weight: 396.26
indicates its normalized occurrence ratio
Number of HBA: 7
(NOR). A high NOR indicates a more
Number of HBD: 0
frequently reported site of metabolism in the
MolLogS : -1.10 (in Log(moles/L)) 31431.52
occurrence ratio does not indicate how likely
AMERICAN JOURNAL OF UNDERGRADUATE RESEARCH
VOL. 11, NOS. 3 & 4 (2012-13)
Molecule ID
cLogP value
Solubility
Molecular
Drug Score
Likeness
Prototype drug, Artemisinin
Table 3. OSIRIS Property explorer values of designed Artemisinin analogues.
Molecule
Potency DBS
Potency DBS
Potency DBS
ic Potency
ed Daily Dose
Table 4. Predicted toxicity of Artemisinin analogues using in-silico lazar toxicity prediction tool
http://lazar.in-silico.de/predict
a molecule is to be metabolised, but rather
c. Phase I Metabolic site prediction using
MetaPrint2D by setting the strictness of
occurring at a particular site in the molecule,
the fingerprint matching in "DEFAULT"
assuming it is metabolised.
and selecting model "ALL (Metabolite
All the designed drug like molecules
taking the pharmacophore of lamivudine are
found to have more drug score than that of
The color highlighting an atom
the prototype drug lamivudine which leads to
indicates its normalized occurrence ratio
confirm that all newly drug like molecules
(NOR). A high NOR indicates a more
might have increased and more potent
frequently reported site of metabolism in the
activity than that of lamivudine.
occurrence ratio does not indicate how likely
AMERICAN JOURNAL OF UNDERGRADUATE RESEARCH
VOL. 11, NOS. 3 & 4 (2012-13)
a molecule is to be metabolised, but rather
occurring at a particular site in the molecule, assuming it is metabolized. (See p. 39 for the Results Color Scheme.)
Figure 8c. Predicted Phase I metabolic site
of AMM3.
All designed Artemisinin analogues
have been predicted as non-carcinogen as well as non-mutagenic by the internet based lazar toxicity prediction tool this confirms that the rational and effective drug like molecules have been designed by SAR and pharmacophore study.
V. DESIGN OF SOME LAMIVUDINE
DERIVATIVES AS POTENT ANTI-RETROVIRAL AGENTS
Lamivudine is a potent nucleoside
analogue reverse transcriptase inhibitor [18].
Figure 8a. Predicted phase I metabolic site
To increase the potency of lamivudine as
anti-HIV agent the –R part of lamivudine
(Fig. 9) is substituted to get more potent designed
increased drug likeness score in MolSoft drug likeness and property explorer tool. The prototype drug lamivudine has a drug likeness score of 1.05 and the designed drug like molecules have greater drug likeness score that of lamivudine
Figure 8b. Predicted Phase I metabolic site
Figure 9. The pharmacophore of lamivudine.
AMERICAN JOURNAL OF UNDERGRADUATE RESEARCH
VOL. 11, NOS. 3 & 4 (2012-13)
Molecule ID
cLogP value
Solubility
Molecular
Drug Likeness
Drug Score
Table 5. OSIRIS Property explorer values of designed Lamivudine derivatives.
a. List of designed Lamivudine Analogues
with MolSoft Drug likeness Scores and
Predicted Molecular Properties
Drug-likeness score of LMA2: 1.23
Molecular Properties and Drug-likeness: Molecular formula: C10 H15 N3 O3 S Molecular weight: 257.08
Number of HBA: 5
Number of HBD: 1
Drug-likeness score of LMA1: 1.16
MolLogS : -1.81 (in Log(moles/L)) 3975.81
Molecular Properties of LMA1:
Molecular formula: C10 H15 N3 O3 S
MolPSA : 53.16 A2
Molecular weight: 257.08
MolVol : 259.27 A3
Number of HBA: 5
Number of stereo centers: 2
Number of HBD: 2
MolLogS : -2.12 (in Log(moles/L)) 1955.97 (in mg/L) MolPSA : 62.24 A2 MolVol : 256.18 A3 Number of stereo centers: 2
AMERICAN JOURNAL OF UNDERGRADUATE RESEARCH
VOL. 11, NOS. 3 & 4 (2012-13)
Figure 9a. Predicted Phase I metabolic site.
of LMA1
Molecule ID:LMA3 Drug-likeness score of LMA3: 1.30 Molecular Properties and Drug-likeness: Molecular formula: C11 H15 N3 O3 S Molecular weight: 269.08 Number of HBA: 5 Number of HBD: 2 MolLogP : 0.17 MolLogS : -2.15 (in Log(moles/L)) 1885.47 (in mg/L) MolPSA : 62.37 A2 MolVol : 272.08 A3
All the designed drug like molecules
taking the pharmacophore of lamivudine are
found to have more drug score than that of
Figure 9b. Predicted Phase I metabolic site
the prototype drug lamivudine which leads to
confirm that all newly drug like molecules
might have increased and more potent
activity than that of lamivudine. b. Phase I Metabolic Site Prediction using
Metaprint2d by Setting the Strictness of the Fingerprint Matching in "DEFAULT" and Selecting Model "ALL (Metabolite 2010.2)"
The color highlighting an atom
indicates its normalized occurrence ratio (NOR). A high NOR indicates a more frequently reported site of metabolism in the metabolite
occurrence ratio does not indicate how likely a molecule is to be metabolized, but rather the
occurring at a particular site in the molecule,
assuming it is metabolized.
Figure 9c. Predicted Phase I metabolic site
AMERICAN JOURNAL OF UNDERGRADUATE RESEARCH
VOL. 11, NOS. 3 & 4 (2012-13)
Molecule
c Potency
Potency DBS
Potency DBS
Potency DBS
Dose mmol
Table 6. Predicted toxicity of Artemisinin analogues using in-silico lazar toxicity prediction tool
http://lazar.in-silico.de/predict
From predicted lazar toxicity it is
likeness, toxicity and other drug like and
clear that the most effective design drug like
molecular properties are easy to handle and
molecule is LMA2 which posses lower
user friendly. Designing of these drug like
predicted toxicity level as compare with
molecules by the pharmacophore study and
prediction of drug like properties of these
lamivudine. But all other design molecules
molecules by the internet based tools hope
will have be more effective than that of
to speed up basic drug design research as
lamivudine as LMA1 and LMA3 as both of
all these tools are user friendly. Currently
these drugs have lower predicted lazar
work is going on the development of a
toxicity than that of lamivudine (as compared
"Miracle Molecule" by the use of internet
by the "confidence" value generated by the
based drug design tools which can be act as
confidence value of predicted toxicity of
anticancer, antibacterial agent as it will
LMA1 and LMA3 than that of prototype drug
contain all the pharmacophores necessary
lamivudine suggests that they are less toxic
for these activities and the work will be
than lamivudine).
reported in near future.
ACKNOWLEDGEMENTS
These entire designed drug like
I am very much thankful to all my friends of
molecules designed on the basis of SAR
Birla Institute of Technology, Mesra, Ranchi-
and pharmacophore study predicted to be
835215 for their healthy criticism and
more effective and potent in nature than that
constant encouragement.
of all protype molecules. The predicted
metabolic sites will be beneficial for
computational chemists for docking analysis of these drugs and in choosing suitable
1. Marshall GR (1987) Computer-Aided
targets. The internet based tools used in
this study for calculating drug score, drug
Pharmacology and Toxicology, Vol. 27:
AMERICAN JOURNAL OF UNDERGRADUATE RESEARCH
VOL. 11, NOS. 3 & 4 (2012-13)
193-213, DOI: 10.1146/annurev.pa. 27.
Rev 46 pp 3–26. DOI:10.1016/S0169-
409X(00)00129-0.
2. Chen, G. S. and Chern, J.-W. (2006)
8. http://www.molsoft.com/mprop/
Computer-Aided Drug Design, in Drug
9. JME courtesy of Dr. Peter Ertl, Novartis
Discovery Research: New Frontiers in
Pharma AG, Basel, Switzerland
the Post-Genomic Era (ed Z. Huang),
10. http://lazar.in-silico.de/predict
John Wiley & Sons, Inc., Hoboken, NJ,
USA. DOI: 10.1002/9780470131862.ch4
3. Ooms F(2000) Molecular Modelling and
12. http://www-metaprint2d.ch.cam.ac.uk/
Computer Aided Drug Design. Examples
13. Chloroquine.
Wikipedia, The Free Encyclopedia.
Chemistry, Current Medicinal Chemistry,
Retrieved 07:34, April 26, 2012, from
Vol. 7(2) 141-158, DOI: http://dx.doi.org
4. Guner, O.F.(2002) History and Evolution
14. Martin RE, Marchetti RV, Cowan AI et
of the Pharmacophore Concept in
"Chloroquine
Computer-Aided Drug Design, Current
transport via the malaria parasite's
Topics in Medicinal Chemistry, Vol.
transporter".
Science 325(5948) pp1680-2.
http://dx.doi.org/
15. Bourne SA, De Villiers K, Egan TJ
(2006). "Three 4-aminoquinolines of
5. Lu IL, Mahindroo N, Liang PH, Peng YH,
antimalarial interest" Acta Crystallogr C
Kuo CJ, Tsai KC, Hsieh HP, Chao YS,
Wu SY.(2006) Structure-based drug
design and structural biology study of
16. Artemisinin. In Wikipedia, The Free
novel nonpeptide inhibitors of severe
Encyclopedia. Retrieved 07:37, April 26,
acute respiratory syndrome coronavirus
main protease, J Med Chem., 49(17) pp
6. Güner OF, ed. (1999). Pharmacophore
17. Noedl H, Se Y, Schaecher K, Smith BL,
perception, development, and use in
Socheat D, Fukuda MM (December
drug design. LaJolla, CA: International
2008). "Evidence of artemisinin-resistant
University Line. ISBN 0-9636817-6-1.
malaria in western Cambodia". N. Engl.
7. C.A. Lipinski; F. Lombardo; B.W.
and P.J. Feeney (2001).
"Experimental
approaches to estimate solubility and
Wikipedia, The Free Encyclopedia.
permeability in drug discovery and
Retrieved 07:41, April 26, 2012, from
development settings". Adv Drug Del
Source: http://www.ajur.uni.edu/v11n3-4/Bhakat%20pp%2035-48.pdf
Final Report DEVELOPING THE ADULT LEARNING SECTOR Lot 2: Financing the Adult Learning Sector (Contract EAC 2012-0073) Prepared for the European Commission/DG Education and Culture FiBS – Forschungsinstitut für Bildungs- und Sozialökonomie DIE – Deutsches Institut für Erwachsenenbildung Berlin, August 27, 2013
SATISFACCION Y PERDIDAS INTERMENSTRUALES CON EL USO CONSECUTIVO DE DISPOSITIVOS INTRAUTERINOS LIBERADORES DE LEVONORGESTREL Helsinki, Finlandia El uso consecutivo de dispositivos intrauterinos liberadores de levonorgestrel se asocia con una reducción de los días de sangrado y pérdidas intermenstruales, y con altos niveles de satisfacción respecto del tratamiento. Human Reproduction 25(6):1423-1427 Jun, 2010 Autores: Heikinheimo O, Inki P, Kunz M, Gemzell-Danielsson G Institución/es participante/s en la investigación: Helsinki University Central Hospital Título original: Predictors of Bleeding and User Satisfaction During Consecutive Use of the Levonorgestrel-Releasing Intrauterine System Título en castellano: Predictores de Hemorragia y Satisfacción de las Usuarias Durante el Uso Consecutivo de Dispositivos Intrauterinos Liberadores de Levonorgestrel Extensión del Resumen-SIIC en castellano: 2.64 páginas impresas en papel A4 Introducción El dispositivo intrauterino (DIU) liberador de levonorgestrel (LNG-DIU) se emplea en Finlandia desde 1990, actualmente se comercializa en más de 120 países y es uno de los dispositivos más usados, junto con el DIU con cobre, entre las mujeres europeas. La duración aprobada de uso es de 5 años. Entre aquellas mujeres que usan LNG-DIU, aproximadamente el 25% se encuentra utilizando su segundo dispositivo consecutivo. El uso del LNG-DIU se asocia con una alta tasa de amenorrea, que alcanza el 60%; en un ensayo clínico, este dispositivo mostró una alta tasa de continuidad, y reducción del sangrado o mantenimiento de la amenorrea durante el primer año de uso del dispositivo por segunda vez consecutiva. El uso de LNG-DIU se caracteriza por un patrón de sangrado irregular y pérdidas intermenstruales durante los primeros meses, que posteriormente disminuyen, a la vez que aumenta la tasa de amenorrea. Sin embargo, no se conocen con certeza los factores predictivos de este tipo de patrón de sangrado asociado al LNG-DIU; en un estudio, se determinó que existía una relación entre la presencia de oligomenorrea al cabo de un año de uso de LNG-DIU y factores tales como la duración basal de la menstruación inferior a los cinco días, el uso del dispositivo como método anticonceptivo y no como tratamiento de la menorragia, y la ausencia de menorragia previa. Sin embargo, no se han estudiado los factores que influyen en el patrón de sangrado durante el uso consecutivo de LNG-DIU. Este estudio tuvo por objetivo analizar los factores predictivos del patrón de sangrado durante el primer año de uso de un segundo LNG-DIU, en mujeres que habían empleado el primer LNG-DIU como anticonceptivo o como tratamiento de la menorragia.