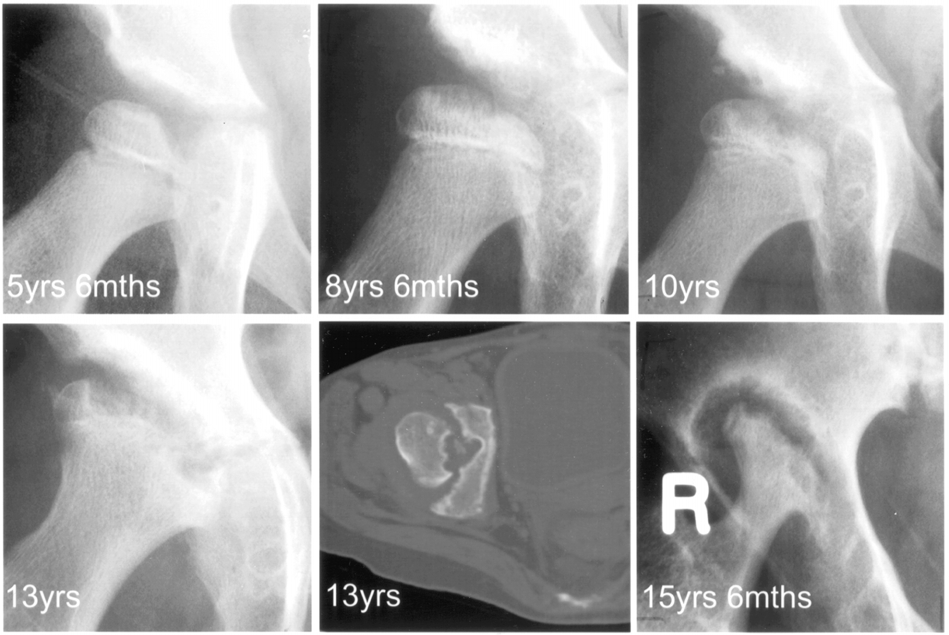
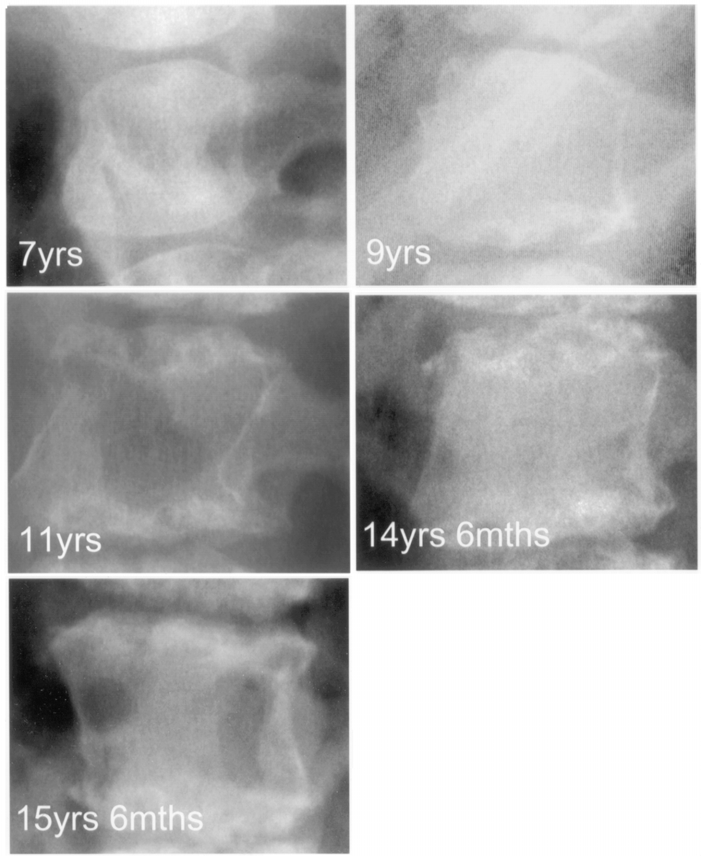
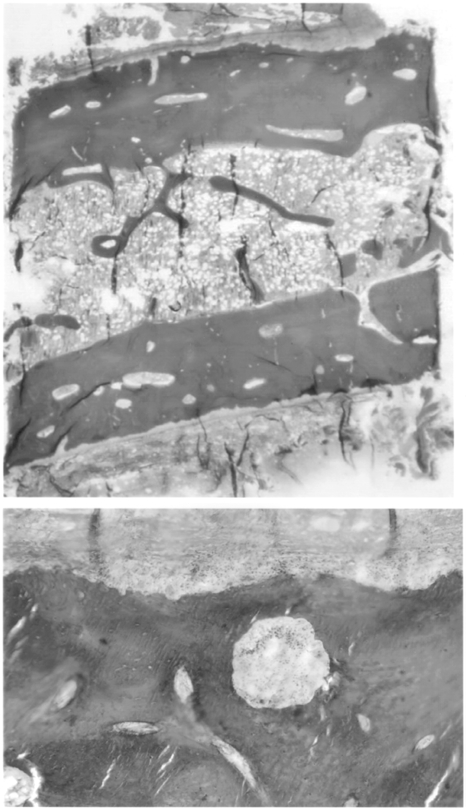
Slide
Phair, Yasmin S. and Miller, Paul K. (2015) Inhibition of pain during the rehabilitation of knee ligament injuries: an interpretive phenomenological analysis. In: BASES Student Conference, 31 March - 1 April 2015, Liverpool John Moore's University, UK. Usage of any items from the University of Cumbria Repository ‘Insight' must conform to the following fair usage guidelines: