Kamagra gibt es auch als Kautabletten, die sich schneller auflösen als normale Pillen. Manche Patienten empfinden das als angenehmer. Wer sich informieren will, findet Hinweise unter kamagra kautabletten.
Mpasmb-hamburg.mpg.de
THE JOURNAL OF BIOLOGICAL CHEMISTRY
Vol. 279, No. 53, Issue of December 31, pp. 55833–55839, 2004
2004 by The American Society for Biochemistry and Molecular Biology, Inc.
Printed in U.S.A.
Hyperphosphorylation and Aggregation of Tau in Experimental
Autoimmune Encephalomyelitis*
Received for publication, August 30, 2004, and in revised form, October 6, 2004
Published, JBC Papers in Press, October 19, 2004, DOI 10.1074/jbc.M409954200
Anja Schneiderद, Gilda Wright Arau
´ jo‡¶, Katarina Trajkovic‡§, Martin M. Herrmann‡,
Doron Merkler储
, Eva-Maria Mandelkow**, Robert Weissert‡¶,‡‡ and Mikael Simons‡§¶§§
From the ‡
Department of Neurology, Hertie Institute for Clinical Brain Research, University of Tu
¨ bingen, Hoppe-Seyler-
Str. 3, 72076 Tu
¨ bingen, Germany, the §
Center for Biochemistry and Molecular Cell Biology, University of Go¨ttingen,
Humboldtallee 23, 37075 Go¨ttingen, Germany, the **
Max-Planck-Unit for Structural Molecular Biology, Notkestr. 85,22607 Hamburg, Germany, and the 储
Department of Neuropathology, University of Go¨ttingen, Robert-Koch-Str. 40,37099 Go¨ttingen, Germany
Axonal damage is a major morphological correlate
(1, 2). There is increasing evidence that axonal damage is the
and cause of permanent neurological deficits in patients
major morphological correlate of permanent neurological defi-
with multiple sclerosis (MS), a multifocal, inflammatory
cits in patients with MS (3, 4). Studies using magnetic reso-
and demyelinating disease of the central nervous sys-
nance spectroscopy/imaging have suggested that neurodegen-
tem. Hyperphosphorylation and pathological aggrega-
eration starts already at the onset of the disease (5).
tion of microtubule-associated protein tau is a common
Accordingly, neuropathological studies revealed significant ax-
feature of many neurodegenerative diseases with ax-
onal injury in early disease stages (6 –9). The finding that
onal degeneration including Alzheimer's disease. We
intralesional axonal damage is related to the degree of inflam-
have therefore analyzed tau phosphorylation, solubility
mation in the lesions has led to the conclusion that inflamma-
and distribution in the brainstem of rats with experi-
tion is not only responsible for demyelination but also for ax-
mental autoimmune encephalomyelitis (EAE), an ani-
onal injury (10). Axonal degeneration in MS is defined by
mal model of MS. Tau was hyperphosphorylated at sev-
characteristic morphological signs such as axonal swellings
eral sites also phosphorylated in Alzheimer's disease
and spheroids (6 –9). Histopathological studies have shown
and became partially detergent-insoluble in EAE brains.
Morphological examination demonstrated accumula-
that axonal damage in MS is associated with axonal accumu-
tion of amorphous deposits of abnormally phosphoryl-
lation of amyloid precursor protein (APP), which is transported
ated tau in the cell body and axons of neurons within
in a kinesin-dependent fashion, indicating impairment of ax-
demyelinating plaques. Hyperphosphorylation of tau
onal transport (6). Because neurons are highly elongated cells,
was accompanied by up-regulation of p25, an activator
their function depends on efficient transport of proteins and
of cyclin-dependent kinase 5. Phosphorylation of tau,
organelles toward synapses. A disturbance in axonal transport
activation of cdk5, and axonal pathology were signifi-
would therefore cause energy depletion at synapses, eventually
cantly reduced when diseased rats were treated with
leading to complete transsection and degeneration of axons in
prednisolone, a standard therapy of acute relapses in
MS. The underlying molecular mechanisms of transport im-
MS. Hyperphosphorylation of tau was not observed in a
pairment and axonal degeneration in MS are so far elusive.
genetic or nutritional model of axonal degeneration or
Axonal degeneration is also found in neurodegenerative dis-
demyelination, suggesting that inflammation as de-
eases (
i.e. Alzheimer's disease, progressive supranuclear palsy,
tected in the brains of rats with EAE is the specific
frontotemporal dementia linked to parkinsonism), which are
trigger of tau pathology. In summary, our data provide
characterized by pathological hyperphosphorylation and as-
evidence that axonal damage in EAE and possibly MS is
sembly of microtubule-associated protein tau into paired heli-
linked to tau pathology.
cal filaments (11, 12). The physiological function of tau is tobind to and stabilize microtubules in a phosphorylation-de-pendent way (11). In addition, tau is involved in regulation of
Multiple sclerosis (MS)1 is an inflammatory disease that
anterograde axonal transport by influencing the attachment/
leads to the destruction of myelin in the central nervous system
detachment rate of molecular motors along microtubules (13).
Pathological hyperphosphorylation of tau as seen in Alzhei-mer's disease causes detachment of tau from microtubules that
* This work was supported by Deutsche Forschungsgemeinschaft
might lead to microtubule breakdown and disruption of axonal
Grants Si 746, SFB 523, and We 1947. The costs of publication of thisarticle were defrayed in part by the payment of page charges. This
transport (loss of function). An imbalance of kinases and phos-
article must therefore be hereby marked "
advertisement" in accordance
phatases has been proposed to contribute to the pathogenesis of
with 18 U.S.C. Section 1734 solely to indicate this fact.
diseases with paired helical filaments (14). A common hypoth-
¶ These authors contributed equally to this work.
esis holds that tau hyperphosphorylation and subsequent de-
‡‡ To whom correspondence may be addressed: Dept. of Neurology,
Hertie Institute for Clinical Brain Research, University of Tu
tachment increases the pool of unbound tau beyond a critical
Hoppe-Seyler-Str. 3, 72076 Tu
¨ bingen, Germany. Tel.: 49-7071-2982141;
concentration, thereby initiating its aggregation into paired
Fax: 49-7071-600137; E-mail:
[email protected].
helical filaments (gain of toxic function) (11, 12, 15, 16).
§§ To whom correspondence may be addressed: Center for Biochem-
istry and Molecular Cell Biology, University of Go¨ttingen, Humbold-allee 23, 37075 Go¨ttingen, Germany. Tel.: 49-551-3899533; Fax: 49-551-3899753; E-mail:
[email protected].
MARK, microtubule-affinity regulating kinase; MAPK, mitogen-acti-
1 The abbreviations used are: MS, multiple sclerosis; APP, amyloid
precursor protein; cdk5, cyclin-dependent kinase 5; EAE, experimental
PP2A, protein phosphatase 2A; SP/TP motif, serin-proline/threonin-
autoimmune encephalitis; GSK-3, glycogen synthase kinase 3;
proline; CNP, 2⬘,3⬘-cyclic nucleotide 3⬘-phosphodiesterase.
This paper is available on line at http://www.jbc.org
Tau Pathology in EAE
In light of the conspicuous axonal abnormalities in MS, we
wondered whether tau abnormalities contribute to neuronaldysfunction and degeneration in experimental autoimmune en-cephalomyelities (EAE), an animal model of MS. Myelin-oligo-dendrocyte-glycoprotein (MOG)-induced EAE in rats resemblesmany characteristic features of MS including multifocal in-flammation, demyelination, and axonal loss. Therefore, wecharacterized tau phosphorylation, solubility, and distributionin rats with acute brainstem EAE.
EXPERIMENTAL PROCEDURES
Induction of EAE—EAE was induced in female LEW.1N rats by
intradermal injection of 50 g rat MOG in saline emulsified (1:1) withcomplete Freund's adjuvant (Sigma) containing 200 g of Mycobacte-rium tuberculosis (strain H 37 RA; Difco Laboratories, Detroit, MI).
Control rats were injected with complete Freund's adjuvant alone. Rats
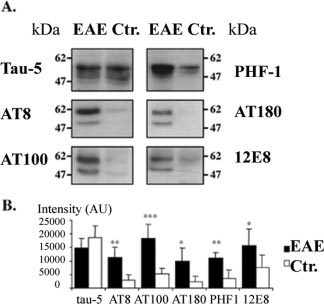
FIG. 1. Hyperphosphorylation of tau in rats with EAE. A, pro-
were scored for clinical signs of EAE and weighed daily. Rats were
tein lysates prepared from EAE brainstems and controls containing
sacrificed 12–13 days after sensitization. The experiments were ap-
equal amounts of proteins were subjected to Western blot analysis with
proved by the regional ethics board.
phosphorylation dependent antibodies PHF-1 (pS396/pS404), AT-8
Treatment of Animals—Rats were treated intraperitoneally with
(pS202/pT205), AT-100 (pT212/pS214), AT-180 (pT231/pS235), and
prednisolone (20 mg/kg) starting on day 8 after sensitization as de-
12E8 (pS262) in addition to the pan-tau antibody tau-5. B, quantitative
scribed (17). 8-Week-old C57/Bl6 mice (n ⫽ 5) were fed with 0.2% (w/w)
analysis of tau expression and phosphorylation of tau epitopes in EAE
cuprizone (bis-cyclohexanone oxaldihydrazone) (Sigma) in ground
and control brains. Values are mean ⫾ S.D., n ⫽ 5 for each value. Three
breeder chow for 5 weeks. Subsequently, brains were snap-frozen in
independent experiments showed similar results (*, p ⬍ 0.05; **, p ⬍
liquid nitrogen for further biochemical analysis (see below). Brains from
0.01; ***, p ⬍ 0.001).
age-matched animals (n ⫽ 5) maintained on a normal diet servedas controls.
nied with extensive axonal damage in these animals (9). In
Western Blotting—Brainstems of LEW.1N rats were dissected, snap-
frozen in liquid nitrogen, and weighted. Tissue was homogenized in
addition, the focal course of the disease with reproducible se-
ice-cold lysis buffer (10 mM Tris-HCl, 150 mM NaCl, 20 mM NaF, 1 mM
vere brainstem pathology facilitates the selection and biochem-
M EGTA, 0.5% Triton X-100, and 0.1% SDS) and protease
ical analysis of lesions. To study expression levels and phos-
inhibitor mixture (Complete, Roche Diagnostics). The homogenates
phorylation status of tau in inflammatory demyelinating
were centrifuged and the protein content in the supernatant was de-
lesions, LEW.1N rats were immunized with 50 g of recombi-
termined. The supernatants were subjected to immunoblotting analy-
nant MOG in complete Freund's adjuvant and brainstems were
sis. As a phosphorylation-independent monoclonal antibody Tau-5 (BDPharmingen) was used. Monoclonal antibodies directed against phos-
dissected at day 12–13 post-immunization. Immunoblotting
phorylated tau epitopes were AT-8, AT-100, AT-180 (Innogenetics,
analysis with the pan-tau antibody tau-5 did not reveal any
Gent, Belgium), 12E8 (P. Seubert, Elan Pharmaceuticals, South San
significant changes in the expression levels of brainstem tau
Francisco, CA), TG-3 and PHF-1 (kindly provided by P. Davies, New
between rats with EAE, adjuvant only immunized controls, and
York). The rabbit polyclonal anti-phospho-MAPK, rabbit polyclonal an-
naı¨ve rats (Fig. 1). To determine whether tau phosphorylation
ti-phospho-independent-MAPK, monoclonal anti-phospho-GSK-3, and
was altered, we used a panel of different phosphorylation-de-
polyclonal phosphorylation-independent GSK-3 were from New Eng-land Biolabs (Beverly, MA). Rabbit anti-p35 antibody was from Santa
pendent antibodies that are commonly used to detect Alzhei-
Cruz Biotechnology (Santa Cruz, CA). Immunoblots were measured
mer tau. One can broadly distinguish two classes of paired
using Scion Image software. Statistical differences were determined
helical filament tau phosphorylation sites (Table I): 1) Ser-Pro
with Student's t test.
and Thr-Pro (SP/TP) sites in the flanking region of tau phos-
Isolation of Insoluble Tau—Brainstems were homogenized in lysis
phorylated by proline-directed kinases such as glycogen syn-
buffer (10 mM Tris-HCl, 150 mM NaCl, 20 mM NaF, 1 mM Na VO , 2 m
thase kinase 3 (GSK-3), cyclin-dependent kinase 5 (cdk5), or
EGTA, 0.5% Triton X-100, and 0.1% SDS) and protease inhibitor mix-ture and centrifuged twice at 10,000 ⫻ g for 5 min. The supernatant was
mitogen-activated protein (MAP) kinase. 2) KXGS motifs in the
removed and recentrifuged at 100,000 ⫻ g for 30 min. The resulting
repeat region phosphorylated by nonproline-directed kinases
pellet was re-extracted with 70% formic acid to recover the insoluble
like microtubule-affinity regulating kinase (MARK/Par-1) and
material. For isolation of crude paired helical filaments (18) brainstem
protein kinase A. Tau phosphorylation at KXGS sites has been
were homogenized in a buffer containing 10 mM Tris-HCl, 0.8 M NaCl,
implicated in the loss of microtubule binding, whereas phos-
1 mM EGTA, 10% sucrose and protease inhibitors, and centrifuged at
phorylation of the flanking region has only a minor effect on
27,000 ⫻ g for 20 min. The pellet was washed once. Both supernatantswere combined, adjusted to 1% Sarkosyl, and incubated for 1 h at 37 °C,
tau microtubule interaction. Replicate blots were stained with
followed by centrifugation at 100,000 ⫻ g for 35 min. The resulting
PHF-1 (pS396/pS404), AT-8 (pS202/pT205), AT100 (pT212/
pellet containing crude insoluble tau was resuspended in 8 M urea.
pS214), and AT-180 (pT231/pS235) to detect distinct proline-
Immunohistochemistry—Immunohistochemistry was performed on
directed kinase phosphorylation sites in the flanking region of
adjacent serial sections of paraffin-embedded brainstem sections using
tau. We observed a dramatic increase of immunoreactivity
standard streptavidin-biotin-peroxidase methods or immunofluores-
toward the PHF-1, AT-8, AT-100, and AT-180 epitopes in
cent dyes. Primary antibodies were used against the following targets:APP, monoclonal antibody 22C11 (Chemicon); phosphorylated tau
brainstem lysates prepared of rats with EAE compared with
(AT-8, AT-100, PHF-1, TG-3); and p35/p25 regulatory subunit of cdk5.
those from control animals (Fig. 1). The average increase was
Bielschowsky silver, Gallyas silver, hematoxylin-eosin, and luxol fast
⬃3.5-fold for PHF-1, ⬃4-fold for AT-8, ⬃4-fold for AT-100, and
blue stainings were done using standard methods.
⬃4.5-fold for AT-180. Next we assessed the phosphorylation
Protein Phosphatase Activity Measurements—Phosphatase activity
status of tau at KXGS motifs in the repeat region phosphoryl-
was determined by phosphatase assay V2460 according to the manu-
ated by nonproline-directed kinases. Among these phosphoryl-
facturer's protocol (Promega).
ation sites, Ser-262 is of particular interest, because its phos-
phorylation is increased in Alzheimer's disease, virtually
Hyperphosphorylation of Tau in Rats with EAE—To study
abolishes tau binding to microtubules and disrupts microtu-
the mechanisms of axonal pathology in MS, we used LEW.1N
bule stability (20, 21). Immunoblotting revealed that phospho-
rats with MOG-induced hyperacute EAE as an experimental
rylation at the 12EA (pS262) epitope was strongly increased in
model (19). As in acute MS, active demyelination is accompa-
rats with EAE (Fig. 1).
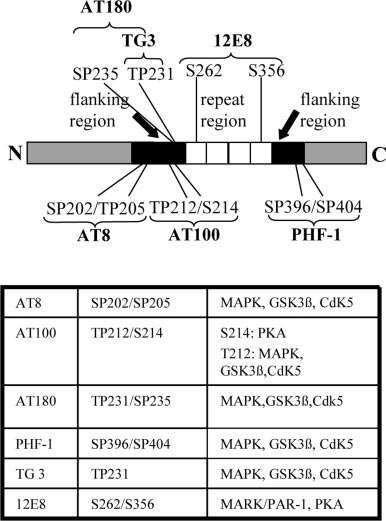
Tau Pathology in EAE
Bar diagram of the longest tau isoform tau40 and its
The repeat region (white bar) consists of 4 repetitive protein se-
quences and is required for microtubule binding. Phosphorylation of itsKXGS motifs leads to detachment of tau from microtubules. In contrast,the phosphorylation of SP/TP motifs in the flanking regions (black bars)have only a modulatory effect on tau-microtubule binding. The tableshows phosphoepitopes of tau together with kinases, identified to phos-phorylate these sites in vivo and in vitro.
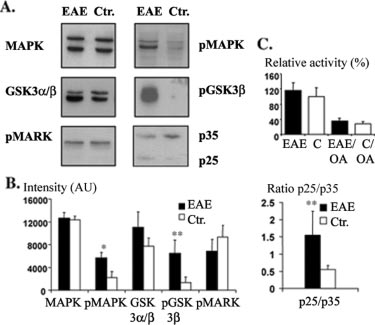
FIG. 2. Analysis of kinase and phosphatase activation in rats
with EAE. A, Western blot analysis of brainstem lysates from EAE rats
and controls with antibodies against various tau-directed kinases (sam-
ples contained equal amounts of total protein). Note that p-MAPK and
p-MARK are directed against active kinase, whereas p-GSK-3 recog-
nizes the inactive enzyme. B, quantitative analysis of the expression
levels of total and activated kinases. Total expression levels of studied
kinases remained unchanged. We observed activation of MAPK, de-
creased activity of GSK-3, no significant change in MARK/Par-1 ac-
tivity, and increased p25/p35 ratio, indicating aberrant activation of
cdk5. Values are mean ⫾ S.D., n ⫽ 5 for each value. Two independent
experiments showed similar results (*, p ⬍ 0.05; **, p ⬍ 0.01). C,
phosphatase activities in brainstem homogenates of rats with and with-
out EAE were determined in duplicate samples using a colorimetric
assay in PP2A specific reaction buffer. PP2A activity is shown in % in
relation to a phosphate standard curve. The assay was also run in the
presence of the phosphatase inhibitor, okadaic acid (OA). The values
are mean ⫾ S.D., n ⫽ 5 for each value. Two independent experiments
showed similar results and no significant difference in phosphatase
activity between EAE and control animals.
phatase, PP2A, is not changed (Fig. 2C).
Hyperphosphorylated Tau and p25/p35 in Degenerating Neu-
rons of Rats with EAE—Previous neuropathological studies
Activation of MAPK and p25/cdk5 in EAE—To determine the
have demonstrated axonal dilatations and spheroids in rats
molecular mechanisms for increased tau phosphorylation, the
with MOG-induced EAE as well as in human MS brains (6 –9).
activity of several known tau-directed protein kinases, includ-
It has been shown recently that damaged axons and spheroids
ing MAPK, GSK-3, cdk5, and MARK were studied (Fig. 2, A
stain strongly positive with an antibody against APP (6).
and B). Immunoblot analysis using an anti-phospho MAPK
Whereas physiological levels of axonal APP are not detected by
(Erk1/2) antibody, which recognizes only the activated form of
this method, APP accumulation is found in damaged axons
MAPK, showed a ⬃2.5-fold increase in MAPK activity in EAE
possibly because of failure of axonal transport (6). We detected
brain lysates compared with that in adjuvant alone immunized
prominent APP staining in dilated axons and spheroids on
or naı¨ve rats. The overall expression levels of MAPK were not
paraffin-embedded brainstems of LEW.1N rats with EAE (Fig.
changed. To assess the activity of cdk5, we determined the
3A). To analyze whether pathologically hyperphosphorylated
levels of cdk5 activators, p35 and its proteolytic fragment p25.
tau was localized in neurons with axonal injury, we performed
The proteolytic conversion of p35 to p25 has been implicated in
immunohistochemical stainings with PHF-1, AT-8, and 12E8
aberrant cdk5 activation leading to tau hyperphosphorylation,
antibodies. All three antibodies prominently stained axons in
cytoskeleton disruption, and neuronal death (22–25). There
EAE brains, particularly dilated and irregularly shaped axons
was a striking increase in the p25/p35 ratio in EAE brainstems,
and axonal spheroids (Fig. 3; and data not shown). In contrast,
suggesting conversion of p35 to p25 in EAE. The activity of the
only weak immunoreactivity of AT-8, PHF-1, and 12E8 was
tau-directed kinase GSK-3 is negatively regulated by phos-
observed in adjuvant-immunized control rats. We next evalu-
phorylation at Ser-9. We found increased phosphorylation at
ated the immunoreactivity with TG-3, an antibody that recog-
Ser-9, whereas levels of total GSK-3␣/ remained unchanged,
nizes phosphorylated tau (pT231) with abnormal conformation
indicating that GSK-3 is not involved in the cascade leading to
and detects early stages of paired helical filaments (29, 30).
tau hyperphosphorylation in EAE. Furthermore, we observed
TG-3 displayed extensive staining of abnormal axons and sphe-
that the activity of the non-proline-directed kinase, MARK,
roids in rats with EAE, indicating a pathological conformation
which phosphorylates Ser-262, was not altered in EAE.
shift of tau (Fig. 3). Similar results were observed when the
In addition to kinase activation, inactivation of phosphatases
conformation-dependent antibody MC-1 was used (data not
can result in hyperphosphorylation of tau (26, 27). We therefore
shown). In addition, an altered compartmentalization of tau
determined the activity of the major tau-directed phosphatase,
with accumulation of amorphous and granular tau deposits in
PP2A, in duplicate samples by a colorimetric assay using a
the soma of neurons within demyelinating plaques was de-
phosphopeptide substrate in the PP2A-specific reaction buffer
tected by PHF-1 and Bielschowsky stainings (Fig. 3B). Previ-
(27, 28). The peptide was dephosphorylated to the same extent
ous studies have shown that the TG-3, AT-8, and PHF-1
by homogenates obtained from rats with EAE compared with
epitopes can be generated in vivo by cdk5 (31, 32). Because we
controls, indicating that the activity of the tau-directed phos-
found a conversion of p35 to p25, indicating an activation of
Tau Pathology in EAE
FIG. 3. Hyperphosphorylated tau and p25/p35 in degenerating
neurons of rats with EAE. A, immunocytochemistry for APP and the
tau-specific phosphoepitopes PHF-1 and TG-3 shows injured axons and
spheroids in cross-sections of brainstems from rats with EAE. B, amor-
phous protein aggregations in the soma of neurons in demyelinating
plaques, stained by Bielschowsky or with PHF-1 antibody. C, axonal
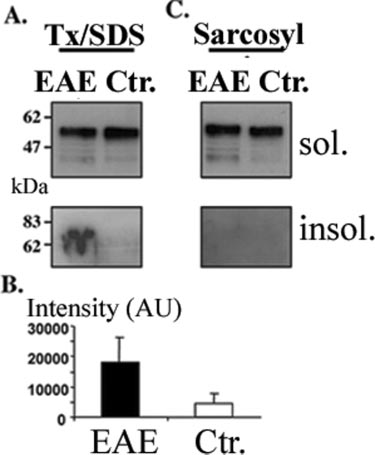
FIG. 4. Tau insolubility and aggregation, but no formation of
swellings reveal prominent staining with an antibody directed against
paired helical filaments in rats with EAE. A, Western blot analysis
the regulatory subunits p25/p35 of cdk5 and phosphorylated tau
(with phosphorylation-independent antibody tau-5) of 0.5% Triton
(PHF1). D, no co-localization of pMAPK and PHF-1, indicating that
X-100, 0.1% SDS (Tx/SDS) soluble and insoluble fractions of brainstem
up-regulation of pMAPK is most likely caused by inflammatory cells.
protein homogenates. Tau becomes partly Tx/SDS insoluble in EAE. B,
Scale bars: 10 m in A, and 25 m in B.
quantitative analysis of Tx/SDS-insoluble tau. Values are mean ⫾ S.D.,n ⫽ 4 for each value (*, p ⬍ 0.05). Two independent experiments showed
cdk5 toward tau phosphorylation in EAE brains, we analyzed
similar results. C, Sarkosyl-soluble and -insoluble fractions, detected
the localization of the regulatory subunits of cdk5 in rats with
with pan-tau antibody tau-5. The insoluble (crude paired helical fila-
EAE. Immunostaining with a C-terminal p35 antibody recog-
ment) fraction of both EAE and control samples did not contain tau.
nizing both p35 and p25 revealed intense staining of axons withabnormal profiles and hyperphosphorylated tau (Fig. 3C). In
pected, the severity of the disease, measured by the clinical
contrast, we did not detect any significant co-localization of
score of each animal, and the number of inflammatory infil-
pMAPK and PHF-1 (Fig. 3D). Taken together, these results
trates were significantly reduced in rats that had been treated
indicate that cdk5, but not MAPK is involved in hyperphospho-
with prednisolone. Previous work in a rat model of autoim-
rylation of tau in EAE.
mune optic neuritis has provided evidence that prednisolone
Tau Insolubility and Aggregation, but No Formation of
treatment may induce apoptosis in retinal ganglia cells (17).
Paired Helical Filaments in Rats with EAE—The accumulation
We therefore evaluated the extent of axonal damage by
of hyperphosphorylated tau in dilated axons and spheroids as
Bielschowsky silver staining on brainstem cross-sections of
well as its pathological conformation, which is known to pre-
rats with EAE. However, signs of axonal injury such as dilated
cede formation of paired helical filaments, is suggestive of
axons and spheroids were virtually absent in the brainstem of
axonal tau aggregations. Because paired helical filaments are
rats that had been treated with prednisolone.
highly insoluble, we analyzed the insolubility of tau in 0.1%
Next, we analyzed the effect of prednisolone treatment on
SDS and 0.5% Triton X-100. EAE brainstems were extracted in
tau phosphorylation. By immunoblotting brainstem lysates
lysis buffer, and the detergent-soluble and -insoluble fractions
with PHF-1 we found a significant reduction (⬃50%) of tau
were subjected to immunoblotting analysis. Whereas tau was
phosphorylation in treated animals compared with untreated
barely detectable in the detergent-insoluble fraction of control
controls (Fig. 5). In addition, treatment with prednisolone re-
brains we found significant amounts of insoluble tau in EAE
duced levels of phosphorylated MAPK and p25 significantly.
brainstems (Fig. 4). The overall levels of tau did not differ
Tau Is Not Hyperphosphorylated in Other Conditions with
between EAE and controls. These data show that in addition to
Primary Oligodendroglial Dysfunction—The causal relation-
hyperphosphorylation, tau also partially forms detergent-insol-
ship of inflammation, demyelination, and axonal degeneration
uble aggregates in EAE. An established biochemical method for
has been difficult to determine in EAE. We therefore analyzed
isolation of paired helical filaments is to take advantage of
the extent of tau phosphorylation in a model of axonal damage
their insolubility in 1% Sarkosyl (18, 33). In contrast to its
that is caused by oligodendroglial dysfunction and not by in-
insolubility in 0.1% SDS and 0.5% Triton X-100, tau from
flammation. Mice that are deficient for the oligodendroglial
diseased animals was soluble in 1% Sarkosyl. Taken together,
protein, 2⬘,3⬘-cyclic nucleotide 3⬘-phosphodiesterase (CNP), de-
these findings demonstrate aggregation of tau without the
velop a progressive neurodegenerative disorder in the absence
formation of paired helical filaments.
of demyelination or inflammation, starting 4 months after
Prednisolone Treatment Reduces Kinase Activation and Tau
birth and are characterized by signs of axonal damage as ob-
Phosphorylation in Rats with EAE—High-dosage prednisolone
served in MS (Fig. 6C and Ref. 34). The development of axonal
treatment is the standard therapy regime in acute relapses of
pathology in CNP-deficient mice was not accompanied by an
MS (1, 2). We therefore tested whether the treatment with
increase in tau phosphorylation (Fig. 6A). To analyze whether
prednisolone could inhibit the pathological cascade leading to
acute demyelination per se can induce tau pathology in axons,
hyperphosphorylation and aggregation of tau in EAE rats. Rats
we fed mice with the demyelinating toxin cuprizone for 5
were treated with 20 mg/kg prednisolone intraperitoneally
weeks. Although cuprizone treatment leads to extensive demy-
from day 8 post-immunization to day 12. At day 12 animals
elination (Fig. 6D and Ref. 35), we did not detect any changes
from treated and untreated groups were sacrificed. As ex-
in tau phosphorylation (Fig. 6B). These data suggest that hy-
Tau Pathology in EAE
FIG. 6. Tau is not hyperphosphorylated in CNP-deficient or
cuprizone-fed mice. A, protein lysates prepared from brains of CNP-
deficient and cuprizone-fed mice containing equal amounts of proteins
were subjected to Western blot analysis with phosphorylation-depend-
ent antibodies PHF-1 and the pan-tau antibody tau-5. B, quantitative
analysis of tau expression and phosphorylation of tau epitopes is shown.
Values are mean ⫾ S.D., n ⫽ 5 (Cuprizone) and n ⫽ 3 (CNP⫺/⫺) for
each value. C, Gallyas staining, revealing prominent axonal pathology
(axonal spheroids, dilated axons) in adult CNP⫺/⫺ mice. D, demyeli-
nation in cuprizone-treated mice. Scale bar: 200 m in D.
the repeat region (e.g. Ser-262) has led to the hypothesis thattau hyperphosphorylation leads to a release of tau from micro-tubules, followed by microtubule breakdown and transport de-cay (loss of function). In addition, hyperphosphorylated taucould be neurotoxic by itself or in its aggregated form (gain oftoxic function). Consistent with the loss of function hypothesiswe found significant tau phosphorylation at Ser-262, one sitewithin the repeat domain that strongly inhibits microtubulebinding and causes detachment of tau from microtubules whenphosphorylated. It is therefore possible that hyperphosphoryl-
FIG. 5. Prednisolone treatment reduces kinase activation and
ation of tau during acute inflammation disengages tau from
tau phosphorylation in rats with EAE. A, clinical score of rats
microtubules, causing their destabilization and impairment of
immunized with MOG and treated with prednisolone from day 8 after
axonal flow. Furthermore, we observed significant abnormal
immunization or left untreated. Quantitative analysis of the effect ofprednisolone treatment. Values are mean ⫾ 0.5 S.D., n ⫽ 5 for each
phosphorylation of SP/TP motifs in the flanking regions of tau
value. B, Bielschowsky silver staining and HE staining of cross-sections
such as PHF-1 (pS396/pS404), AT-8 (pS202/pT205), AT-180
from prednisolone-treated or untreated rats with EAE. Note the virtual
(pT231/pS235), and AT-100 (pT212/pS214) epitopes.
absence of inflammation and axon degeneration in prednisolone-treated
To identify downstream kinases involved in the sequential
animals. Scale bar: 10 m. C, Western blot analysis of tau phosphoryl-ation (PHF-1 epitope) in prednisolone-treated and untreated rats with
toxic hyperphosphorylation of tau at SP/TP motifs we per-
EAE. Phosphorylation of the PHF-1 epitope is decreased in prednisolo-
formed immunoblotting with several activity dependent anti-
ne-treated animals. Whereas total amounts of MAPK are unchanged,
bodies against active MAPK, inactive GSK-3, and upstream
active MAPK is decreased and the ratio of cdk5 activating subunits
activators of cdk5 because all of these have been described to
p25/p35 is shifted toward p35 in prednisolone-treated animals. D, quan-titative analysis of the effect of prednisolone treatment. Values are
phosphorylate tau in vivo and in vitro. In contrast to GSK-3,
mean ⫾ S.D., n ⫽ 5 for value (*, p ⬍ 0.05).
which was found to be down-regulated we detected an increasein MAPK activity as well as an increase in the ratio of cdk5
perphosphorylation of tau is part of a specific pathway trig-
activators p25/p35. Considering that MAPK is also expressed
gered by an inflammatory attack in the central nervous system
in astrocytes and microglial cells it is possible that the detected
of rats with EAE.
activated MAPK could originate from glial cells rather thanneurons (36). Indeed, we did not detect significant co-localiza-
tion of pMAPK and PHF-1 in EAE. In contrast, cdk5 and its
Here we show that 1) tau derived out of EAE brains is
activators p25/p35 are primarily expressed in neurons (see also
abnormally hyperphosphorylated at sites that define tau pa-
Fig. 3C) and have been localized and purified from brain mi-
thology in Alzheimer's disease; 2) that hyperphosphorylation is
crotubules (37, 38). In addition, transgenic mice overexpressing
most likely because of an activation of cdk5 rather than to
p25/cdk5 display increased tau phosphorylation at SP/TP mo-
decreased phosphatase activity; 3) that pathologically hyper-
tifs in the flanking region as well as tau aggregation and
phosphorylated tau is localized in injured neurons of EAE
neurodegeneration (23–25). Cdk5 is recruited to the neuronal
brains; and 4) that tau becomes partially insoluble and under-
membrane by its interaction with membrane-anchored p35
goes a conformational shift that is thought to precede paired
(39). Aberrant activation of cdk5 occurs when the myristoy-
helical filament formation, whereas aggregation into paired
lated domain of p35 is cleaved to p25, leading to release of
helical filaments is not observed.
p25-bound cdk5 into the cytoplasmic compartment. Several
These findings raise the question of how tau could mediate
lines of evidence indicate that only p25 but not p35-bound cdk5
the axonal damage that occurs in active MS lesions. The fact
phosphorylates tau in vitro, thereby inducing pathological al-
that the binding of tau is regulated by phosphorylation within
terations in neurons (22–25, 40, 41).
Tau Pathology in EAE
It is interesting to note that conversion of p35 to p25 is
in MS and EAE lesions correlates with the extent of inflamma-
regulated by the calcium-dependent cysteine protease calpain
tion, suggesting that the primary insult is an inflammatory
as activation of calpain together with impaired calcium home-
attack (6 –9). However, the causal relationship of inflamma-
ostasis has been observed in EAE and MS brains (42, 43). It is
tion, demyelination, and axonal degeneration has been difficult
therefore tempting to speculate that increased calcium influx
to determine. To address this question we used a genetic model
might trigger a cascade of pathological events that lead to
of axonal damage, which is triggered by oligodendroglial dys-
calpain activation, followed by conversion of p35 to p25 and
function instead of inflammation. CNP-deficient mice display
increased and pathological tau phosphorylation at epitopes
axonal loss in the absence of demyelination or inflammation.
such as PHF-1, AT-8, and AT-100.
Although the axonal degeneration observed in this model dis-
It has generally been assumed that filamentous tau aggre-
plays similar ultrastructural features such as APP positive
gations as they are described in Alzheimer's disease, fronto-
axonal swellings and spheroids, hyperphosphorylation of tau is
temporal dementias and other tauopathies might be direct
only found in EAE, but not in CNP-deficient mice. In addition,
mediators of neuronal toxicity because the clinical progression
cuprizone-induced demyelination did not lead to tau pathology
of Alzheimer's disease correlates with distribution and amount
in axons. These data indicate that demyelination per se is not
of tau aggregates (44, 45). Tau is a highly soluble protein
sufficient to induce changes in tau phosphorylation. Further-
because its sequence consists mostly of hydrophilic residues.
more, one can speculate that inflammation as seen in EAE
The exact molecular mechanisms of its abnormal aggregation
triggers a specific pathway of axonal damage, which is distinct
into paired or straight helical filaments are not completely
from the one caused by oligodendroglial dysfunction or demy-
understood. In vitro studies propose extrinsic (polyanions, ox-
elination. This raises the question to which extent axonal dam-
idative environment) as well as intrinsic factors (increased tau
age can be reversed by reducing the inflammatory load and
concentration, tau mutations that promote -structure) as pos-
how steroid treatment that is the standard therapy for acute
sible reasons for aggregation (46). Hyperphosphorylation has
relapsing MS influences axonal damage. Our findings suggest
also been assumed to cause pathological aggregation of tau.
that a prednisolone pulse treatment during the active phase of
The antibody TG-3 recognizes a conformation-dependent
inflammation does not only reduce the amount of inflammatory
epitope that has been reported to precede paired helical fila-
infiltrates, but also the extent of axonal damage.
ment formation (29). Because we observed a marked TG-3 and
In summary our data provide evidence that axonal damage in
MC-1 staining of injured axons, we tested tau solubility and
EAE is associated with tau hyperphosphorylation and aggrega-
aggregation in the EAE model. We could identify one fraction of
tion. These pathological tau alterations can be partially pre-vented by early prednisolone treatment. These findings are of
tau that had become insoluble in 0.5% Triton X-100, 0.1% SDS,
particular relevance because the amount of axonal damage is a
indicating the formation of tau aggregates. Amorphous aggre-
major determinant of persistent neurological deficits in MS pa-
gates in the soma of neurons and axonal spheroids that are
tients. Our results might open new perspectives for understand-
stained by PHF-1 and Bielschowsky silver impregnation could
ing molecular pathology and treatment of multiple sclerosis.
be the morphological correlate of the 0.5% Triton X-100, 0.1%SDS-insoluble tau fraction. A widely accepted biochemical
Acknowledgments—We are grateful to P. Davies and P. Seubert for
method to isolate paired helical filaments of Alzheimer brains
the generous gift of tau antibodies and K.-A. Nave for providing CNP-
is based on their insolubility in 1% Sarkosyl. However, no tau
deficient mice. We thank K. de Graaf for assistance with perfusions ofanimals.
reactivity could be detected in the Sarkosyl-insoluble fractionof EAE brainstem lysates. In line with the absence of Sarkosyl-
insoluble tau aggregates, no paired helical filaments were ob-
1. Compston, A., and Coles, A. (2002) Lancet 359, 1221–1231
served by Gallyas or Bielschowsky staining. Insolubility and
2. Steinman, L., Martin, R., Bernard, C., Conlon, P., and Oksenberg, J. R. (2002)
therefore aggregation of tau in the absence of paired helical
Annu. Rev. Neurosci. 25, 491–505
3. Bjartmar, C., and Trapp, B. D. (2001) Curr. Opin. Neurol. 14, 271–278
filaments raises the question of the relevance of these findings
4. Kornek, B., and Lassmann, H. (1999) Brain Pathol. 9, 651– 656
for axon degeneration in EAE. It is poorly understood whether
5. Matthews, P. M., De Stefano, N., Narayanan, S., Francis, G. S., Wolinsky,
J. S., Antel, J. P., and Arnold, D. L. (1998) Semin. Neurol. 18, 327–336
hyperphosphorylated tau is toxic by itself or oligomeric tau
6. Ferguson, B., Matyszak, M. K., Esiri, M. M., and Perry, V. H. (1997) Brain 120,
aggregations or stable fibrils are required to cause cellular
7. Trapp, B. D., Peterson, J., Ransohoff, R. M., Rudick, R., Mork, S., and Bo, L.
dysfunction. Evidence is now accumulating that formation of
(1998) N. Engl. J. Med. 338, 278 –285
paired helical filaments is not necessary for tau toxicity. Phos-
8. Kuhlmann, T., Lingfeld, G., Bitsch, A., Schuchardt, J., and Bru¨ck, W. (2002)
phorylation of tau in a temporally ordered series can already
Brain 125, 2202–2212
9. Kornek, B., Storch, M. K., Weissert, R., Wallstroem, E., Stefferl, A., Olsson, T.,
cause neurodegeneration in the absence of aggregation (47).
Linington C., Schmidbauer, M., and Lassmann, H. (2000) Am. J. Pathol.
Furthermore, it has been shown that misfolded and oligomer-
10. Neumann, H. (2003) Curr. Opin. Neurol. 16, 267–273
ized proteins can cause cellular dysfunction before the deposi-
11. Lee, V. M., Goedert, M., and Trojanowski, J. Q. (2001) Annu. Rev. Neurosci. 24,
tion of stable fibrils (48). Small protein assemblies that can
only be detected biochemically and not by imaging techniques
12. Goedert, M., Spillantini, M. G., and Davies, S. W. (1998) Curr. Opin. Neuro-
biol. 8, 619 – 632
might be in part responsible for cytotoxicity. For example, in
13. Ebneth, A., Godemann, R., Stamer, K., Illenberger, S., Trinczek, B., and
mice expressing mutant human APP cell death occurs well
Mandelkow, E. (1998) J. Cell Biol. 143, 777–794
14. Bue´e, L., Bussie re, T., Bue´e-Scherrer, V., Delacourte, A., and Hof, P. R. (2000)
before A amyloid deposition (49, 50). Similarly, several lines
Brain Res. Rev. 33, 95–130
of evidence suggest that tau-related neurodegeneration can
15. Grundke-Iqbal, I., Iqbal, K., Quinlan, M., Tung, Y. C., Zaidi, M. S., and
Wisniewski, H. M. (1986) J. Biol. Chem. 261, 6084 – 6089
occur without or precede paired helical filament formation (47,
16. Friedhoff, P., von Bergen, M., Mandelkow, E. M., and Mandelkow, E. (2000)
51). Whether the biochemically detected tau aggregates in EAE
Biochim. Biophys. Acta 1502, 122–132
brainstems represent toxic intermediates of the fibrillogenic
17. Diem, R., Hobom, M., Maier, K., Weissert, R., Storch, M. K., Meyer, R., and
Ba¨hr, M. (2003) J. Neurosci. 23, 6993–7000
process remains to be established. It is also possible that de-
18. Lee, V. M., Wang, J., and Trojanowski, J. Q. (1999) Methods Enzymol. 309,
tergent-insoluble tau is part of larger poorly soluble cellular
19. Weissert, R., Wallstroem, E., Storch, M. K., Stefferl, A., Lorentzen J., Lass-
aggregates (for example, found in axonal spheroids) formed as
mann, H., Linington, C., and Olsson, T. (1998) J. Clin. Investig. 102,
a consequence of impaired axonal flow and are thus not related
to any fibrillogenic process.
20. Seubert, P., Mawal-Dewan, M., Barbour, R., Jakes, R., Goedert, M., Johnson,
G. V., Litersky, J. M., Schenk, D., Lieberburg, I., Trojanowski, J. Q., and
Neuropathological studies have shown that axonal damage
Lee, V. M.-Y. (1995) J. Biol. Chem. 270, 18917–18922
Tau Pathology in EAE
21. Biernat, J., Gustke, N., Drewes, G., Mandelkow, E.-M., and Mandelkow, E.
34. Lappe-Siefke, C., Goebbels, S., Gravel, M., Nicksch, E., Lee, J., Braun, P. E.,
(1993) Neuron 11, 153–163
Griffiths, I. R., and Nave, K. A. (2003) Nat. Genet. 33, 366 –374
22. Patrick, G. N., Zuckerberg, L., Nikolic, M., de la Monte, S., Dikkes, P., and
35. Matsushima, G. K., and Morell, P. (2001) Brain Pathol. 11, 107–116
Tsai, L. H. (1999) Nature 402, 615– 622
36. Shin, T., Ahn, M., Jung, K., Heo, S., Kim, D., Jee, Y., Lim, Y. K., and Yeo, E. J.
23. Noble, W., Olm, V., Takata, K., Casey, E., Mary, O., Meyerson, J., Gaynor, K.,
(2003) J. Neuroimmunol. 140, 118 –125
LaFrancois, J., Wang, L., Kondo, T., Davies, P., Burns, M., Veeranna,
37. Ishiguro, K., Takamatsu, M., Tomizawa, K., Omori, A., Takahashi, M., Arioka,
Nixon, R., Dickson, D., Matsuoka, Y., Ahlijanian, M., Lau, L. F., and Duff,
M., Uchida, T., and Imahori, K. (1992) J. Biol. Chem. 267, 10897–10901
K. (2003) Neuron 38, 555–565
38. Sobue, K., Agarwal-Mawal, A., Li, W., Sun, W., Miura, Y., and Paudel, H. K.
24. Cruz, J. C., Tseng, H.-C., Goldman, J. A., Shih, H., and Tsai, L.-H. (2003)
(2000) J. Biol. Chem. 275, 16673–16680
Neuron 40, 471– 483
39. Dhavan, R., and Tsai, L.-H. (2001) Nat. Rev. Mol. Cell. Biol. 2, 749 –759
25. Ahlijanian, M. K., Barrezueta, N. X., Williams, R. D., Jakowski, A., Kowsz,
40. Van den Haute, C., Spittaels, K., Van Dorpe, J., Lasrado, R., Vandezande, K.,
K. P., McCarthy, S., Coskran, T., Carlo, A., Seymour, P. A., Burkhardt,
Laenen, I., Geerts, H., and Van Leuven, F. (2001) Neurobiol. Dis. 8, 32– 44
J. E., Nelson, R. B., and McNeish, J. D. (2000) Proc. Natl. Acad. Sci. U. S. A.
41. Hashiguchi, M., Saito, T., Hisanaga, S.-I., and Hashiguchi, T. (2002) J. Biol.
97, 2910 –2915
Chem. 277, 44525– 44530
26. Gong, C. X., Shaikh, S., Wang, J. Z., Zaidi, T., Grundke-Iqbal, I., and Iqbal, K.
42. Shields, D. C., and Banik, N. L. (1999) J. Neurosci. Res. 1, 533–541
(1995) J. Neurochem. 65, 732–738
43. Nitsch, R., Pohl, E. E., Smorodchenko, A., Infante-Duarte, C., Aktas, O., and
27. Goedert, M., Jakes, R., Qi, Z., Wang, J. H., and Cohen, P. (1995) J. Neurochem.
Zipp, F. (2004) J. Neurosci. 24, 2458 –2464
65, 2804 –2807
44. Braak, H., and Braak, E. (1991) Acta Neuropathol. 82, 239 –259
28. Kins, S., Crameri, A., Evans, D. R., Hemmings, B. A., Nitsch, R. M., and Go¨tz,
J. (2001) J. Biol. Chem. 276, 38193–38200
45. Arriagada, P. V., Growdon, J. H., Hedley-Whyte, E. T., and Hyman, B. T.
29. Jicha, G. A., Lane, E., Vincent, I., Otvos, L., Jr., Hoffmann, R., and Davies, P.
(1992) Neurology. 42, 631– 639
(1997) J. Neurochem. 69, 2087–2095
46. Barghorn, S., and Mandelkow, E. (2002) Biochemistry 41, 14885–14896
30. Augustinack, J. C., Schneider, A., Mandelkow, E.-M., and Hyman, B. T. (2002)
47. Nishimura, I., Yang, Y., and Lu, B. (2004) Cell 116, 671– 682
Acta Neuropathol. 103, 26 –35
48. Selkoe, D. J. (2003) Nature 426, 900 –904
31. Hamdane, M., Sambo, A.-V., Delobel, P., Begard, S., Violleau, A., Delacourte,
49. Hsia, A. Y., Masliah, E., McConlogue, L., Yu, G. Q., Tatsuno, G., Hu, K.,
A., Bertrand, P., Benavides, J., and Buee, L. (2003) J. Biol. Chem. 36,
Kholodenko, D., Malenka, R. C., Nicoll, R. A., and Mucke, L. (1999) Proc.
Natl. Acad. Sci. U. S. A. 96, 3228 –3233
32. Illenberger, S., Drewes, G., Trinczek, B., Biernat, J., Meyer, H. E., Olmsted,
50. Mucke, L., Masliah, E., Yu, G. Q., Mallory, M., Rockenstein, E. M., Tatsuno,
J. B., Mandelkow, E.-M., and Mandelkow, E. (1998) Mol. Biol. Cell 9,
G., Hu, K., Kholodenko, D., Johnson-Wood, K., and McConlogue, L. (2000)
J. Neurosci. 20, 4050 – 4058
33. Greenberg, S. G., and Davies, P. (1990) Proc. Natl. Acad. Sci. U. S. A. 87,
51. Wittmann, C. W., Wszolek, M. F., Shulman, J. M., Salvaterra, P. M., Lewis, J.,
Hutton, M., and Feany, M. B. (2001) Science 293, 711–714
Source: http://www.mpasmb-hamburg.mpg.de/mand-pdf/Schneider__Simons_2004_JBC_Tau-Phos+EAE.pdf
Biowaiver Monographs for Immediate Release Solid OralDosage Forms Based on Biopharmaceutics ClassificationSystem (BCS) Literature Data: Chloroquine Phosphate,Chloroquine Sulfate, and Chloroquine Hydrochloride R.K. VERBEECK,1 H.E. JUNGINGER,2 K.K. MIDHA,3 V.P. SHAH,4 D.M. BARENDS5 1Faculty of Pharmacy, Rhodes University, Grahamstown, South Africa 2Leiden/Amsterdam Center for Drug Research, Leiden University, Division of Pharmaceutical Technology,Leiden, The Netherlands
epidemiology Biostatistics and public Health - 2013, volume 10, number 2 SyStematic reviewS and meta- and pooled analySeS A systematic review of the cost-effectiveness of lifestyle modification as primary prevention intervention for diabetes mellitus type 2 Katrin I. Radl(1), Carolina Ianuale(2), Stefania Boccia(2) Background: diabetes is one of the leading causes of death, and has a huge economic impact on the burden of society. Lifestyle interventions such as diet, physical activity and weight reducing are proven to be effective in the prevention of diabetes. To encourage policy actions, data on the cost-effectiveness of such strategies of prevention programmes are needed. MeThods: a systematic review of the literature on the cost-effectiveness of prevention strategies focusing on lifestyle interventions for diabetes type 2 patients. a weighted version of drummond checklist was used to further assess the quality of the included studies. resuLTs: six studies met the inclusion criteria and were therefore considered in this paper. Intensive lifestyle intervention to prevent diabetes type 2 is cost-effective in comparison to other interventions. all studies were judged of medium-to-high quality.concLusIons: policy makers should consider the adoption of a prevention strategy focusing on intensive lifestyle changes because they are proven to be either cost-saving or cost-effective.