Kamagra gibt es auch als Kautabletten, die sich schneller auflösen als normale Pillen. Manche Patienten empfinden das als angenehmer. Wer sich informieren will, findet Hinweise unter kamagra kautabletten.
Mtsaj.co.za2
Medical Technology SA
Volume 25 No. 1 June 2011
Peer reviewed
rEviEW
PATHOGENESIS AND FUTURE TREATMENTS OF SYSTEMIC LUPUS
ERYTHEMATOSUS: THE ROLE OF CYTOKINES AND ANTI-CYTOKINES?
W. J. Maule
University of Johannesburg, Department of Biomedical Technology, Faculty of Health Sciences, South Africa.
email: [email protected] tel: +27 (0)11 559 6265 fax: +27 (0)11 559 6558
cytokine production and cytokine levels in patients with slE
Systemic lupus erythematosus (SLE) is a chronic multisystem
Several cytokines are in involved in the pathogenesis of SLE [11]
autoimmune connective tissue disorder, which has variable
and more than 30 years ago [12] immune interferon (IFN-γ) was
clinical manifestations that range from mild to life-threatening
found in the serum of patients with SLE and showed a good
[1]. These can be characterised by multiple organ damage, very
correlation between (IFN-γ) titres and disease activity.
high titres of autoantibodies and immune complex deposition.
T-helper cells 1 (Th1) cytokines such as IFN-γ, IL-12, and
Interestingly the former of these characteristics may precede
T-helper cells 2 (Th2) cytokines IL-4, IL-6 and IL-10 are each
the clinical manifestations of SLE by many years [1]. It is well
considered to play a role in the course of human SLE [13, 14].
recognised that the probable influence of oestrogen hormonal
Other proinflammatory cytokines such as IL-1, IL-17, IL-23 and
effect in women during childbearing years increases their chances
tumour necrosis factor alpha (TNFα) [15] are also involved along
of developing SLE by 10-15 times [2-5]. The immunopathogenic
with these Th1 and Th2 cytokines.
hallmark of SLE is the polyclonal B cell activation, which leads to hyperglobulinanaemia, autoantibody production and immune complexes. All of these factors contribute to the conventional belief that SLE is a disease primarily of these autoantibodies and immune complex deposition, the latter contributing to inflammation by virtue of complement activation and the engagement of complement and fragment crystallisable (Fc) – receptors [6, 7] ultimately inflicting injury to a variety of organ systems (Figure 1). Mediation of these inflammatory responses is characterised by the influx of various cell populations and also to a large extent by the generation of proinflammatory cytokines. The clinical manifestations in inflammatory diseases such as SLE and rheumatoid arthritis (RA) are thought to be influenced by the balance between proinflammatory and anti-inflammatory cytokines [9].
Cytokines are soluble factors and are mainly
produced by helper T (Th) cells. They also play a crucial role in the differentiation, maturation and activation of various immune cell types [8]. In order to monitor disease activity and predict disease severity certain cytokines may act as biomarkers [9]. Recent work for example, using microarray techniques and genetic analysis has strengthened the association between cytokine dysregulation and SLE [10]. These breakthroughs show some promise in understanding the immunoregulatory networks of autoimmune diseases, which are influenced by multiple factors, particularly in regard to these cytokines and their interactions.
Through systematic review of published literature,
only those cytokines that have significant
involvement in the pathogenesis of SLE in the
‘human model' and those that represent a
relatively easy target for therapeutic intervention
Figure 1: Simplified diagram of the Immunopathogenesis of SLE.
(i.e. the anti-cytokines) will be reviewed.
(adapted from Mok et al
. [93])
ISSN 1011 5528 www.smltsa.org.za
5
Medical Technology SA
Volume 25 No. 1 June2011
Figure 2: IL-6-producing cells and biological activities of IL-6. IL-6 is produced by lymphoid and nonlymphoid cells, such as
T cells, B cells, monocytes, fibroblasts, keratinocytes, endothelial cells, mesangial cells, and several kinds of tumor cell (top
of figure). IL-6 also has a wide range of biological activities on various target cells (bottom of figure).
(reproduced with permission [36])
1. interleukin 6 (il-6)
to stimulate the monocyte/macrophage fraction of PBMCs taken from SLE patients to produce IL-6 [26]. Another interesting
IL-6 is a proinflammatory cytokine which is synthesised
observation was that lymphoblastoid cells that were isolated
principally by monocytes, fibroblasts and endothelial cells
from SLE patients exhibited high levels of IL-6 and blocking IL-
(Figure 2). IL-6 secretion can also be found in both T and
6, which resulted in the inhibition of anti-dsDNA production
B lymphocytes [15] and its production is stimulated by IL-
in vitro [27]
. However, using a widely applied method to study
α, but subdued by IL-4, IL-10 and IL-13. In
combination with type 1 interferons, one of the most important
the activation of the innate immune system i.e. the
in vitro
effects of IL-6 is to activate B lymphocytes, drive plasma-cell
stimulation of whole blood using lipopolysaccharide (LPS), IL-6
differentiation and to augment the immunoglobulin secretion [16,
production was significantly lower in SLE patients as compared
17]. Additionally, IL-6 acts on multipotential progenitor cells, is a
to normal individuals [28].
neutrophil activator and stimulates megakaryocytes to produce
Unlike normal individuals, B lymphocytes from SLE patients
platelets. It also induces terminal macrophage and osteoclast
were found to spontaneously generate large amounts of
differentiation as well as pyrexia and the production of acute
immunoglobulins (Ig). There was however, a significant reduction
phase proteins [36].
in this Ig production when IL-6 was blocked and this production
In total contrast to these proinflammatory effects, IL-6 is also
was only restored after exogenous IL-6 administration [23]. In
involved in a number of unique anti-inflammatory reactions. For
addition these B lymphocytes also secrete anti-double-stranded
example, IL-1 and TNF-α stimulate the synthesis of each other
DNA (anti-ds DNA), with different B lymphocyte populations
as well as IL-6, however, the latter is involved in terminating
contributing to this in a number of different ways. For example,
this reaction as well as being involved in the upregulatory
it was shown that the majority of these autoantibodies were
inflammatory cascade [17].
produced
ex vivo by low density B lymphocytes [29], whereas
The association of IL-6 in the pathogenesis of SLE in humans is
high density B cells had little effect. It was also shown that
still controversial [18] although support for this association has
in response to IL-6, low density B lymphocytes from patients
been published using several murine models [19-21].
with active SLE were capable of directly differentiating into Ig secreting cells [30]. CD5 expression is also down-regulated
1.1. role of il-6 in human slE
by IL-6 via DNA methylation, which promotes activation and
Human SLE patients have been shown to have increased IL-6 [22-
subsequent expansion of auto-reactive B cells seen in SLE
24] levels that are allied to disease activity [23] or anti-DNA levels
patients [30]. The IL-6 abnormalities seen in SLE may well be
[22], in some but not all studies [24].
due, in part to, genetic differences. For example, Linker-Israeli
In one study [25], SLE patients had a significantly higher frequency
et al [31] demonstrated that alleles of the adenosine/tyrosine (AT)
of IL-6 secreting peripheral blood mononuclear cells (PBMCs)
rich minisatellite situated in the 3' region flanking the IL-6 gene,
compared to those of healthy controls. This may well be due to
was associated with SLE patients of either Caucasian or African-
environmental factors as exposure to UV light has been shown
American origin, but not in the control group.
6 www.smltsa.org.za ISSN 1011 5528
Medical Technology SA
Volume 25 No. 1 June 2011
It is well proven that the classical marker auto-antibodies
There was a notable reduction in inflammatory markers, auto-
seen in SLE are anti-ds DNA antibodies and although the titre of
antibody levels and in disease activity (SELENA-SLEDAI from
those antibodies in the serum of SLE patients can be a reflection
9.5 at baseline to 5.5 at 20 weeks) with a median decrease of
of disease activity in lupus nephritis, for example, their exact
38% in the 4 mg/kg dosage group and 56% in the 8 mg/kg
role remains unclear. It has been shown however, that anti-
dosage group. Unfortunately almost all the patients developed
dsDNA can have a direct effect on cytokine expression in a
a significant dose-related neutropenia with concomitant high
variety of cells. They can also upregulate the expression of the
rates of infections [50].
proinflammatory cytokines IL-1 and IL-6 in endothelial cells [32-
Although neutropenia may limit the maximum dosage of
34] and they can stimulate the expression and release of IL-1,
tocilizumab in patients with SLE, the observed clinical and
IL-6, IL-8, IL-10 and TNF [35] (from human resting mononuclear
serological responses are promising and warrant further studies
to establish the optimal dosing regimen and efficacy.
1.2. il-6 and lupus nephritis
2. interleukin 10 (il-10)
IL-6 has been shown in several studies to have proliferative
The cytokine IL-10 is mainly produced by lymphocytes
effects on mesangial cells thereby modulating injury in
and monocytes. It also impedes the activation of antigen
immunologically generated nephritis. Two studies demonstrated
presenting cells (APCs) and down-regulates the expression of
that mesangial proliferation in mesangial proliferative
co-stimulatory molecules such as major histocompatibility
glomerulonephritis correlated well with the urinary IL-6 levels
complex class II (MHC II) and B7 expression [51]. IL-10 also
[37, 38]. Further studies demonstrated high urinary excretion of
inhibits T cell function by diminishing the expression of other
IL-6 in patients with active lupus nephritis. The levels of IL-6
proinflammatory cytokines such as TNFα, IL-1, IL-6, IL-8
were significantly elevated in patients with proliferative lupus
and IL-12 [52, 53]. In addition to these inhibitory functions IL-
nephritis (World Health Organisation (WHO) Class III and IV)
10 boosts B cell mediated proliferation, thereby increasing
with concomitant high titres of anti-dsDNA antibodies [39, 40].
survival, proliferation, differentiation and immunoglobulin class
IL-6 levels were also found to be much higher in patients with
switching, resulting in increased antibody secretion, which
active nephritis as compared to those patients with dormant
promotes the inflammation seen in SLE [54].
renal disease [24, 40]. Additionally it was found that there was
In particular, the production of IL-10 and TNFα, two mutually
enhanced
in situ expression of IL-6 in lupus nephritis, mainly
associated cytokines, play a complex and opposite role in these
along the renal glomeruli and tubles [41-43].
systemic inflammatory responses that has been found to be
Interestingly IL-6 has also been shown to have a positive
deregulated in SLE patients (Figure 3).
association with the Neuropsychiatric syndromes of systemic
All these findings, plus the addition of environmental influences
lupus erythematosus (NPSLE). For example elevated levels of
are suggestive of a combination between genetic and disease-
IL-6 have been reported in the cerebrospinal fluid (CSF) of
induced events. IL-10 and TNFα for example, have been linked
patients with NPSLE, without subsequent damage to the blood-
to SLE and genetic polymorphorisms at the promoter regions of
brain barrier [44-46].
both these genes [55] is associated with their over production,
To summarise, IL-6 has an important role in mediating local
particularly that of IL-10 [56]. However, previous studies of a
inflammation and insults of various tissues.
much larger magnitude which included patient family members with increased IL-10 production [57], failed to confirm this
1.3. therapeutic implications of il-6 in slE
association [58].
As previously stated in a number of studies, IL-6 was found to
Increased IL-10 production might also explain B cell
be elevated in both human and murine lupus [21-24].
hyperactivity and autoantibody production, two of the main
IL-6 released from PBMC for example, directly correlated
indicators of the immune dysregulation seen in SLE.
with disease activity and the treatment response seen in lupus
In line with this; the association between IL-10, disease activity,
nephritis patients [47].
immune complexes isolated from the serum of SLE patients
Other studies have confirmed that there was an increased
as well as monoclonal anti-dsDNA antibodies, induced IL-
expression of the IL-6 agonistic receptor gp130 on peripheral
10 production in healthy monocytes [35, 59]. IL-10 might also
lymphocytes in SLE patients, and that the levels correlated with
regulate dendritic cells (DCs) and T cell function, by promoting
overall disease activity [47, 48].
Th2 deviation of the overall immune response (Figure 3) [60].
Taking this into account it has been suggested that gp130 could
2.1. therapeutic implications of il-10 in slE
be a useful biomarker to monitor both the activity of disease and subsequent treatment responses in those patients [48].
Although one of the major factors is still the absence of a
Using murine models where the success of IL-6 antagonism is
therapeutic agent which is suitable for long-term administration
well proven, a phase 1 dose finding study was set up to evaluate
in human patients with SLE IL-10 was the first cytokine to be
the use of a monoclonal antibody tocilizumab (Anti-IL-6 R
blocked [64], which has led to use of anti-IL-10 antibody in the
Ab) in human SLE patients [49]. A total of sixteen patients with
treatment of this disease [61].
moderately active disease [as defined by the Safety of Estrogens
An over-production of IL-10 has been demonstrated in murine
in Lupus Erythematosus National Assessment (SELENA) and
models of SLE [62]. Using continuous early-onset therapy with an
Systemic Lupus Erythematosus Disease Activity Index (SLEDAI),
anti-IL-10 antibody however, delayed autoimmunity in NZB/W
(i.e. a SELENA-SLEDAI score of between 3 and 10 or active
mice and improved their overall survival rate from 10 to 80%
glomerulonephritis] were given tocilizumab in one of three
[63]. In a pilot study using an anti-IL-10 murine monoclonal
doses (2 mg/kg in 4 patients, 4 mg/kg in 6 patients, and 8 mg/
antibody (MoAb), which neutralizes human IL-10, Llorente
et
kg in 6 patients) twice weekly for 12 weeks. Patients were then
al. [64] evaluated the clinical efficacy and safety of this antibody
monitored for an additional 8 weeks [50].
in a total of six patients with steroid dependent SLE.
ISSN 1011 5528 www.smltsa.org.za
7
Medical Technology SA
Volume 25 No. 1 June2011
Figure 3: Interactions of IL-10 and TNFα
and their role in the development of SLE. This figure represents a simplified model
of the complex relationship between IL-10 and TNFα
in lupus disease. Both cytokines are produced by multiple cells types
of the innate and adaptative immune system, in particular DCs, monocytes/macrophages, (Mø) and specific effector T cells.
Th1 cells produce the proinflammatory cytokine TNFα
which activates DCs and other antigen presenting cells (APCs), and
induces the production of IL-10. In addition, TNFα
promotes inflammation and apoptosis, generating neoantigens that could
result in autoantibody production. On the other hand, IL-10, a Th2 cytokine, antagonise Th1 differentiation and inhibits
APCs and T cells. Conversely, IL-10 is a potent stimulator of B cell proliferation, differentiation and antibody production.
Thus, B cell activation in presence of neo-antigens may lead to autoantibody secretion and immune complexes formation,
thus resulting in tissue damage affecting diverse organs. STAT; signal transducer and activator of transcription.
(reproduced with permission [55])
The treatment consisted of administering 20mg/day of MoAb
of transforming growth factor (TGF-β) and other important
intravenously for a total of 21 consecutive days. The patients
inflammatory cytokines including IL-6, IL-21 and IL-23 [68-70].
were then followed up monthly for a total period of 6 months.
The latter also enhances IL-17 production by memory T cells [71].
The therapy was well tolerated in all six patients and although
These observations strongly suggest that the presence of an
all had significant improvement of their cutaneous lesions
inflammatory signal of some sort is required to transform these
and/or joint symptoms during MoAb administration, they also
naïve CD4 T cells to become pro-inflammatory. The cytokine
developed antibodies against it.
IL-21 for example, was found to influence Th-17 differentiation.
This study not only suggests that the use of MoAb may be of
Unlike IL-6 it is produced by Th17 cells and the T-follicular
benefit in the management of refractory SLE, but that a much
helper cells, but not by APCs. Mangan
et al [70] claimed that
larger, randomized and blinded study using a humanized anti-
one of its functions was that of an auto-amplifier of the Th17
IL-10 MoAb is required. Such an agent might soon be available
response. IL-17 also upregulates the expression of intercellular
adhesion molecule-1 (ICAM-1) through the facilitation of T cell activation and infiltration into tissues [72]. Th17 cells can also
3. interleukin 17 (il-17)
assist as an independent T helper effector cell subset, which
IL-17 is an ancient cytokine, and is a type 1 transmembrane
promote an inflammatory response through cytokine secretion
protein, produced by activated T cells and is intimately related to
(i.e. IL-17A, IL-17F, IL-21 and IL-22) (Figure 4) [73]. In regard
epithelia, especially those of the intestinal mucosa [65, 66]. IL-17 is
to the pathogenesis of SLE, this collection of cytokines can
a potent pro-inflammatory cytokine that also plays an important
stimulate B lymphocytes, to initiate the local inflammation and
role in the immune response against fungi and bacteria [67].
tissue injury often seen in this disease [73].
As stated, IL-17 is produced by activated T lymphocytes, with
Recent studies support and confirm the role of IL-17 in SLE
the ‘Th17' cells being the most energetic producer. Th17 cells
pathogenesis [74, 75].
originate when naïve CD4 T cells are primed in the presence
8 www.smltsa.org.za ISSN 1011 5528
Medical Technology SA
Volume 25 No. 1 June 2011
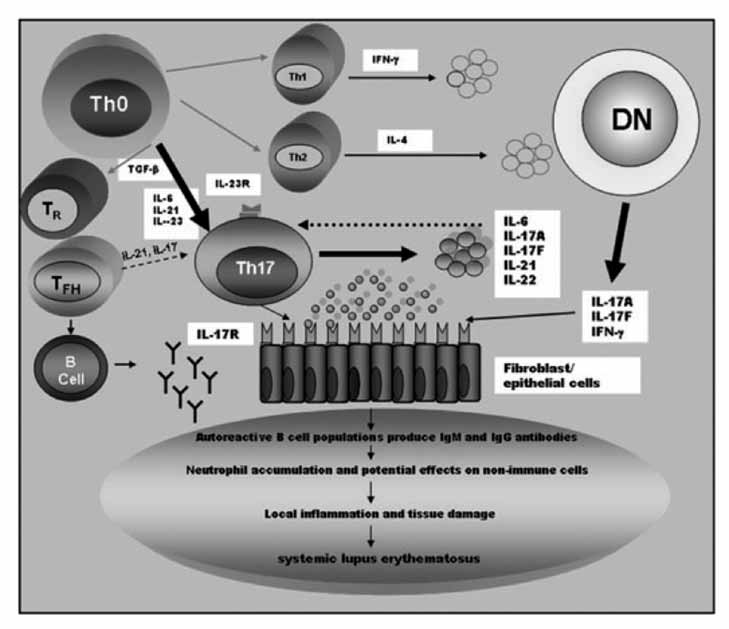
Figure 4: Proposed model for the role of T-helper type 17 (Th17) cells and interleukin (IL)-17
in the pathogenesis of systemic lupus erythematosus (SLE) (reproduced with permission [72]).
CD4+ cells differentiate into Th1, Th2 and Th17 effector cells as well as double-negative (DN) T
cell subsets. The cytokine milieu characteristic of SLE patients (lack IL-2; high levels of IL-6 and
IL-21) could promote Th17 expansion. Th17 cells serve as an independent T helper effector cell
subset promoting inflammation through cytokine secretion. The signature cytokines include IL-
17A, IL-17F, IL-21 and IL-22. These cytokines have stimulatory effects on B cells and activate local
inflammation and tissue damage leading subsequently to the pathogenesis of SLE [72].
3.1. role of il-17 in human slE
Statins which are used extensively in lowering cholesterol in humans have been shown to have immune-modulatory effects
Current evidence suggests that SLE patients have abnormally
and have recently emerged as possible therapeutic agents for
high levels of IL-17 and IL-23 in their serum [75, 76] and that the
autoimmune disease including SLE, although results in animal
level of IL-17 correlates with disease activity [75, 77]. In a recent
models have been both conflicting and controversial [82, 83]. It
study Crispin et al [78] demonstrated that a significant portion
was demonstrated that statins could suppress the secretion of
of IL-17 in SLE patients was derived from double negative
IL-17 by Th17 cells [84] and that they have beneficial effects in
(DN) TCR-αβ+CD4-CD8- T cells. DN T cells represent a small
improving the rate of progression of chronic kidney disease in
subset in healthy individuals, whereas in the peripheral blood
human SLE patients with lupus glomerulonephritis [85].
of SLE patients these cells represent a much larger component producing proinflammatory cytokines including: IL-17, IFN-γ
4. interleukin 23 (il-23)
and IL-1β [78] (see Figure 1). These DN T cells and Th17 cells
IL-23 plays an important role in the development of pathogenic
have also been seen in renal biopsies of patients with lupus
Th17 cells and the subsequent production of IL-17 [86-88] (Figure
nephritis, adding credence to their pathogenic role in renal
IL-23 is a type 1 covalently linked heterodimeric cytokine
As well as its direct proinflammatory activities, IL-17's effects
comprising of p19 and p40 subunits, which are shared with IL-
in other cell types may also contribute to SLE pathogenesis.
12 (Figure 5). IL-23 is produced in the main by both activated
Dong et al [79], for example, observed that there was an increased
dendritic and phagocytic cells [89, 90] and recent studies suggest
production of total IgG, anti-dsDNA IgG and IL-6 by peripheral
that rather than IL-12 it is the most important cytokine for the
blood mononuclear cells of patients with lupus nephritis. All
pathogenesis of autoimmune diseases [91, 92]. IL-23 and IL-12
of these findings indicate that IL-17 may participate in the
share a common p40 subunit, which binds to a common IL-
activation of B cells in patients with SLE.
12 β1 receptor (Figure 5) [94]. Activated/memory T cells, T-cell
This latter finding was verified by the fact that these SLE-
clones and natural killer cell lines in humans, preferentially
derived B cells when cultured in the presence of IL-17 had an
express the IL-23 receptor (IL-23R), which is made up of the IL-
increased production of anti-DNA [79]. Evidence in regard to
23 complex, and a common IL-12 receptor β1.
the importance of this pathological mechanism in human SLE
Because of its central role in the pathogenesis of various
as well as the main question of whether IL-17 blockade (anti-
autoimmune diseases including inflammatory bowel disease,
IL-17) will be therapeutically useful for SLE patients has still to
Duerr et al [95] and ankylosing spondylitis [96, 97], studies focusing
be fully elucidated [80, 81].
on its role in SLE have arisen.
ISSN 1011 5528 www.smltsa.org.za 9
Medical Technology SA
Volume 25 No. 1 June2011
Figure 5: A schematic diagram of the different components making up the IL-12 and IL-23 receptors
and the common STAT4 activation pathway.
IL-12β1 and IL-12β2 each consist of three fibronectin type III and 2 cytokine receptor domains with an
additional immunoglobulin (Ig)-like domain on the latter. IL-23R closely resembles IL-12β2, however
without the fibronectin type III domains. (reproduced with permission [94]).
4.1. role of il-23 in human slE
sample size in different populations, to confirm this association [101].
As stated earlier, cytokine-mediated immunity plays an important role in the pathogenesis of SLE, and implications of
4.2. therapeutic implications of il-23 in slE
this are seen in animal models. Additionally, a number of studies in human SLE have also shown a need to focus on IL-23 and its
At present the majority of the data in regard to IL-23 have come
receptor. Wong et al [75], confirmed that ex vivo syntheses of IL-
from studies using murine models, which may not be wholly
17 by IL-23 or IL-18 produced from co-stimulated lymphocytes
relevant for human SLE. There is however growing evidence that
was much higher in patients with SLE than the control group
in the human model IL-23 plays a role in the development of the
and increased levels of IL-12, IL-17 and Interferon-inducible
disease and that the use of anti-IL-23 therapy to treat the subset
protein-10 (CXCL 10) had both significant and positive
of SLE patients that are characterised by high levels of IL-23 is
correlations with SLEDAI [75]. It was also shown by Huang et
now a distinct possibility [81]. There are two issues that need to be
al [96] that in active SLE patients, the mRNA levels of p19, p40
taken cognisance of in this regard. Firstly, both IL-23 and IL-23R
of PBMC were significantly higher when compared with levels
are critical in mediating antimicrobial defences and in cross-
in their inactive counterparts [96]. In another study, Hillyer et al
regulating other cell subsets, therefore the risk of infectious
[97], reported that in Rheumatoid Arthritis (RA) synovial cultures,
complications need to be taken into account. Secondly most of
IL-23R blockade resulted in a significant inhibition of TNF-α
the data regarding IL-23 have been from murine models.
(57%), IL-1β (51%) and IL-6 (30%) [97]. All of these results
Further comprehensive studies are therefore required, especially
suggest that IL-23 may have pathogenic activity in a proportion
in regard to the therapeutic potential of IL-23 in the treatment
of the patients tested that have late-stage RA. In a recent study,
Kwan et al [98] examined the urinary sediment of three groups of
5. B-lymphocyte stimulators (Blys)
subjects: those with active SLE, with history of lupus nephritis in remission, those with no history of renal involvement SLE
The B-lymphocyte stimulator (BLyS also known as the B cell-
and healthy individuals. In each case they quantified the mRNA
activating factor belonging to the TNF family, or BAFF) [102]
expression of IL-17, IL-23 and other Th 17-related cytokines.
was identified as a novel TNF family ligand almost 10 years
The results concluded that the urinary expression of Th-17
ago [102-105] where it was found to be the key in both the
related genes was increased in the SLE patients when compared
selection and survival of B cells. The expression of the BLyS
to the control group. The degree of this up-regulation however,
protein is confined to myeloid lineage cells (e.g. monocytes,
was inversely proportional to the disease activity [98].
macrophages, dendritic cells and activated neutrophils) [106-108].
This pattern was contradictory to previous studies on the urinary
Although the levels of BLyS are well established and constant,
mRNA expression of Th1- and Th2-related genes [99, 100], which
its expression and secretion can be increased by inflammatory
showed an up-regulation of Th1-related genes and a down-
cytokines, such as IL-2, TNF-α and IFN-γ [107, 109, 110]. BLyS can
regulation of Th2-related genes in patients with SLE, with
bind to three types of receptors: BLyS receptor 3 (also know
the magnitude being proportional to overall disease activity.
as BAFFR), transmembrane activator-1 and calcium modulator
Although these findings suggest a regulatory role of IL-23 in the
and cyclophilin ligand-interactor (TACI) and B cell maturation
pathogenesis of SLE, further studies are required using a larger
antigen (BCMA). It has been shown that BLyS can bind to all
10 www.smltsa.org.za ISSN 1011 5528
Medical Technology SA
Volume 25 No. 1 June 2011
these three receptors on B cells, as opposed to a proliferation-
In a previous study the results showed no over-expression of
inducing ligand (APRIL), which can only engage to TACI and
the type I IFN gene in the blood from SLE patients whereas
BCMA [111]. The most important receptor amongst the three was
an over expression of several other IFN-inducible (IFI) genes
found to be BAFFR as it is was the one that mediated most of
have since been found [131]. This finding was in agreement with
the BLyS effects.
other studies, which demonstrated that peripheral blood from SLE patients had remarkable homogeneous gene expression
5.1. implications for B cells and Blys in human slE
patterns, including an over expression of IFI genes, implying
Elevated serum levels of BLyS protein have been observed in
IFN involvement in SLE [132-136].
patients with autoimmune disease, including those with SLE
Hopefully in the future, genetic mapping may be of assistance in
and these levels correlated with their anti-dsDNA levels [112-115].
predicting the development and severity of the disease and that
In one survey where the serum BLyS levels and disease activities
IFN regulated cytokines may also be used to monitor disease
were measured; healthy controls had normal serum BLyS levels
activity and subsequent organ damage [137-138].
over time, compared to SLE patients who had escalating levels.
The effect of a single dose of anti-IFN monoclonal antibody in
Results displayed a persistent elevation in 25% of patients tested
SLE patients was evaluated in a phase I dose-escalation study
and an intermittent elevation in another 25% of patients [116].
[139]. The results noted a reduction in disease activity where the
These findings suggest that BLyS may figure significantly in the
over expression of IFN-inducible genes were neutralised in a
development of autoimmune disease and in particular SLE, thus
dose-dependent manner. In addition, a number of doses had
making it an ideal target for SLE therapy.
clinical benefit in terms of SLEDAI. Currently there are two phase 2 trials taking place to evaluate the effects of anti-IFN
5.2. therapeutic implications of Blys in slE
monoclonal antibody in SLE patients [140, 141].
There are many conflicting reports regarding BLyS-targeted
7. tumour necrosis factor-a (tnf-α)
therapy using belimumab, a fully human monoclonal antibody (IgG1) that binds to BLyS and inhibits its biological activity. In a
Tumour necrosis factor- alpha (TNF-α) is a proinflammatory
phase I randomised controlled clinical trial [117], the safety and
as well as an immunoregulatory cytokine. It is expressed as
efficacy of belimumab in SLE patients was studied. Although
a homotrimer on the cell surface in a soluble form after the
there was a reduction in CD20+ B cells in this dose-ranging
activation of macrophages and dendritic cells, (Figure 3) with
study as compared to the placebo, there was no significant
divergent effects on the immune system in SLE [142, 143].
improvement in disease activity as assessed by the SELENA-SLEDAI score. In a phase II dose-ranging study [118], three
7.1 role of tnf-α in human slE
different doses of bilimumab were evaluated in SLE patients who
The significance of TNF-α in the pathogenesis of SLE remains
were randomised over a 52 week period and again there was no
controversial as one might argue that it is beneficial and that
significant difference between the combined bilimumab group
TNF blockade would be unfavourable. However, the in vivo
versus the placebo group. A total of 71.5% of the patients were antinuclear antibody (ANA) positive and interestingly in the
data ascertained from a number of SLE patients suggests the
subgroup that were ANA positive the SLEDAI score was reduced
by 29% at week 52. This trial was later continued as an open-
The levels of TNF-α are actually increased in the serum of SLE
label extension study and a four-year safety and efficacy for
patients and are closely correlated with overall disease activity, [144, 145]
some 237 patients has also recently been published [119] where
where an abundant TNF-α expression was demonstrated
serologically active patients had sustained improvement in their
in lupus nephritic kidneys [146, 47].
flares over time. There was also a decline in multiple pathogenic
The beneficial effects of TNF-α blocking therapy have been
antibodies, including anti-dsDNA and anticardiolipin.
shown in a series of studies in patients with other autoimmune
Multi-centre phase 3 trials, of Benlysta™ (belimumab) (BLISS-52
diseases, but the results were conflicting in that these patients
and BLISS-76) in seropositive patients with SLE are currently
developed antinuclear factors, anti-ds DNA and anticardiolipin
being evaluated in two large randomised, double-blind,
antibodies as well as a lupus-like syndrome [147, 148]. All symptoms
placebo-controlled studies [120]. The results of these two pivotal
and autoantibodies disappeared when TNF-α blocking therapy
phase 3 trials, suggest that belimumab can reduce SLE disease
was discontinued.
activity and that it may be the long-awaited new effective
Nevertheless, the findings of elevated serum TNF-α in active
therapy for this disease.
SLE and the over expression of TNF-α in active lupus nephritis [47, 149] provided the rationale for using TNF-α antagonism in SLE
6. type i interferons (type i ifn)
patients [145, 150, 151].
Although type I and type II IFNs have both been implicated
Unfortunately, long term treatment using TNF-α blocking
in the pathogenesis of human SLE [121-125], the type I IFNs
therapy was associated with high rates of serious adverse
are regarded as the most important. For example the initial
reactions [152-155]. For example in two randomised trials [156, 157]
symptoms in many patients with active SLE are often flu-like,
that were designed to evaluate the efficacy and safety of the TNF
where they exhibit symptoms such as fever and fatigue, both of
inhibitors; infliximab and etanercept in SLE patients, both had
which reflect high serum type I IFN levels, which is also relevant
to be terminated prematurely.
to overall disease activity and severity [126-128 ].
Taking all of this recent information into consideration it is
The classical triggers of type I interferon are viral DNA and RNA
highly unlikely that TNF inhibition will be used routinely in the
with signals being mediated via the Toll-like receptors (TLR)
treatment of SLE.
or the retinoic acid inducible gene I (RIG-I) like receptors [129].
8. concluding remarks
Although type I IFNs are manufactured by all leucocytes, the major producer is the cell subset; plasmacytoid dendritic cells
This review has discussed a vast amount of information in
(PDC) in response to TLR7 or TLR9 activation [130].
regard to the pathogenic link between the various cytokines
ISSN 1011 5528 www.smltsa.org.za 11
Medical Technology SA
Volume 25 No. 1 June2011
and SLE (Figure 6) as well as new approaches that target
in achieving rapid disease control and minimise corticosteroid
the pro-inflammatory cytokine pathways which lead to the
use. The role of these agents in the maintenance phase of SLE
amelioration of clinical disease in human SLE. However, the
still remains undefined, and whether the interference of these
elicited inflammatory response characterised by both the influx
events becomes an important therapeutic target will depend
of various cell populations mediated to a large extent by the
on the results of ongoing and continuing clinical trials. Given
generation of these proinflammatory cytokines still needs to
the safety concerns regarding the long-term use of such agents
be fully elucidated. Most of the recent trials have dealt with
especially in SLE, where complications of infection and
the use of agents that target the cytokines involved in this
malignancy may arise, the major challenge for the future will
inflammatory chain of events in the induction phase of severe
be to define which one of these targets will actually be useful in
disease, or in symptoms refractory to conventional treatment
the management of this disease.
such as corticosteroids. They may therefore offer an advantage
Figure 6: Simplified schematic diagram showing the complex interactions between various immune cells and cytokines
which lead to the pathogenesis of SLE. (reproduced with permission [9])
Holers VM. (2003). Complement receptors. In Smolen JS,
Arbuckle MR, Mc Clain MT, Rubertone MV, et al. (2003).
Lipsky PE eds. Targeted therapies in rheumatology. London,
Development of autoantibodies before the clinical onset of
New York: Martin Dunitz: 167-180.
systemic lupus erythematosus. New England Journal of Medi-
Steinman L. (2007). A brief history of T 17, the first major
cine.; 349: 1526-1533.
revision in the T 1/T 2 hypothesis of T cell-mediated tissue
Masi AT, Kaslow A. (1978). Sex effects in systemic lupus ery-
damage. Nature Medicine; 13(2): 139-145.
thematosus: a clue to pathogenesis. Arthritis and Rheuma-
Yap DYH, Neng Lai K. (2010). Cytokines and their roles in
tism; 21(4): 480-484.
the pathogenesis of Systemic Lupus Erythematosus: From ba-
Lahita RG, Bradlow RA, Fishman J, Kunkel HG. (1982). Ab-
sics to recent advances. Journal of Biomedicine and Biotech-
normal estrogen and androgen metabolism in the human with
nology; 2010: Article ID 365083.
systemic lupus erythematosus. American Journal of Kidney
10. Lee H-M, Sugino H, Nishimoto N. (2010). Cytokine networks
Diseases; 2(1): 206-211.
in Systemic Lupus Erythematosus. Journal of Biomedicine and
Rood MJ, Van der Velde EA, Ten Cate R, Breeveldt FC, Huiz-
Biotechnology; 2010: Article ID 676284.
inga TW. (1998). Female sex hormones at the onset of sys-
11. Kunz M, Ibrahim SM. (2009). Cytokines and cytokine pro-
temic lupus erythematosus affect survival. British Journal of
files in human autoimmune diseases and animal models of
Rheumatology; 37(9): 1008-1010.
autoimmunity. Mediators Inflamm.: 979258.
Gubbels Bupp MR Jørgenson TN, Kotzin BL. (2008). Identi-
12. Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL,
fication of candidate genes that influence sex hormone de-
Notkins AL, et al. (1979). Immune interferon in the circu-
pendent disease phenotypes in mouse. Genes and Immunity;
lation of patients with autoimmune disease. New England
Journal of Medicine.; 30: 5-8.
Clynes R, Dumitru C, Ravetch JV. (1998). Uncoupling of im-
13. Horwitz DA, Jacob CO. (1994). The cytokine network in the
mune complex formation and kidney damage in autoimmune
pathogenesis of systemic lupus erythematosus and possible
nephritis. Science; 279: 1072-1074.
therapeutic implications. Springer Seminars in Immunology;
12 www.smltsa.org.za ISSN 1011 5528
Medical Technology SA
Volume 25 No. 1 June 2011
16: 181-200.
cell activation in patients with systemic lupus erythematosus.
14. Dean GS, Tyrrell-Price J, Crawley E, Isenburg DA. (2000).
Clin. Exp. Immunol.; 77: 31-36.
Cytokines and systemic lupus erythematosus. Annals of the
31. Linker-Israeli M, Wallace DJ, Prehn J, Michael D, Honda M,
Rheumatic Diseases; 59: 243-251.
Taylor KD, Paul-Labrador M, Fischel-Ghodsian N, Fraser PA,
15. Hiran T. (1998). IL-6 and its receptor. International Reviews
Klinenberg JR. (1999). Association of IL-6 gene alleles with
of Immunology; 16: 249-284.
systemic lupus erythematosus (SLE) and with elevated IL-6 ex-
16. 16. Schimpl A, Wecker E. (1972). Replacement of T-cell
pression. Genes and Immun.; 1: 45-52
function by a T-cell product. Nature: New Biology; 237(70):
32. Lai KN, Leung JC, Lai KB, Wong KC, Lai CK. (1996). Upregu-
lation of adhesion molecule expression on endothelial cells
17. Thakey E, Lipsky PE, Illei GG. (2004). Rationale for inter-
by anti-DNA autoantibodies in systemic lupus erythematosus.
leukin- 6 blockade in systemic lupus erythematosus. Lupus;
Clinical Immunol. Immunopathol.; 81: 229-238.
13(5): 339-343.
33. Lai KN, Leung JC, Lai KB, Lai FM, Wong KC. (1996). In-
18. Spronk PE, ter Borg EJ, Limberg PC, Kallenberg CG. (1992).
creased release of von Willebrand factor antigen from en-
Plasma concentration of IL-6 in systemic lupus erythemato-
dothelial cells by anti-DNA autoantibodies. Ann. Rheum.
sus; an indicator of disease activity? Clin. Exp. Immunol.; 90:
Dis.; 55: 57-62.
34. Neng LK, Leung JC, Bik LK, Li PK, Lai CK. (1996). Anti-DNA
19. Kobayashi I, Matsuda T, Saito T, et al. (1992). Abnormal dis-
autoantibodies stimulate the release of interleukin-1 and in-
tribution of IL-6 receptor in aged MRL/1pr mice: elevated ex-
terleukin-6 from endothelial cells. J. Pathol.; 178: 451-457.
pression on B cells and absence on CD4+ cells. Int. Immunol.;
35. Sun KH, Yu CL, Tang SJ, Sun GH. (2000). Monoclonal anti-
4: 1407-1412.
double-stranded DNA autoantibody stimulates the expres-
20. Suzuki H, Yasukawa K, Saito T. et al. (1993). Serum soluble
sion and release of IL-I beta, IL-6, IL-8, IL-10 and TNF-alpha
interleukin-6 receptor in MRL/1pr mice is elevated with age
from normal human mononuclear cells involving in the lupus
and mediates the interleukin-6 signal. Eur. J. Immunol.; 23:
pathogenesis. Immunology; 99: 352-360.
36. Naka T, Nishimoto N, Kishimoto T. (2002). The paradigm
21. Tang B, Matsuda T, Akira S, et al. (1991). Age-associated in-
of IL-6: from basic science to medicine. Arthritis Res.; 4(3):
crease in interleukin 6 in MRL/1pr mice. Int. Immunol.; 3:
37. Horii Y, Iwano M, Hirata E, et al. (1993). Role of interleukin-6
22. Grondal G, Gunnarsson I, Ronnelid J, Rogberg S, Klareskog L,
in the progression of mesangial proliferative glomerulonephri-
Lundberg I. (2000). Cytokine production, serum levels and
tis. Kidney Int. Suppl.; 39:S71-S75.
disease activity in systemic lupus erythematosus. Clin. Exp.
38. Iwano M, Dohi K, Hirata E, Horii Y, Shiiki H, Ishikawa
Rheumatol.; 18: 565-570.
H. (1992). Induction of intreleukin 6 sysnthesis in mouse
23. Linker-Israeli M, Deans RJ, Wallace DJ, Prehn J, Ozeri-Chen
glomeruli and cultured mesangial cells. Nephron; 62: 58-65.
T, Klinenberg JR. (1991). Elevated levels of endogenous IL-6
39. Iwano M, Dohi K, Hirata E, et al. (1993). Urinary levels of
in systemic lupus erythematosus. A putative role in pathogen-
IL-6 in patients with active lupus nephritis. Clin. Nephrol.;
esis. J. Immunol.; 147: 117-123.
24. Peterson E, Robertson AD, Emlen W. (1996). Serum and uri-
40. Tsai CY, Wu TH, Yu CL, Lu JY, Tsai YY. (2000). Increased ex-
nary interleukin-6 in systemic lupus erythematosus. Lupus;
cretions of beta2-microglobulin, IL-6 and IL-8 and decreased
excretion of Tamm-Horsfall glycoprotein in urine of patients
25. Hagiwara E, Gourley MF, Lee S, Klinman DK. (1996). Dis-
with active lupus nephritis. Nephron; 85: 207-214.
ease severity in patients with systemic lupus erythematosus
41. Fukatsu A, Matsuo S, Tamai H, Sakamoto N, Matsuda T,
correlates with an increased ratio of interleukin-10-interfer-
Hirano T. (1991). Distributon of interleukin-6 in normal and
on-gamma-secreting cells in the peripheral blood. Arthritis
diseased kidney. Lab. Invest.; 65: 61-66.
Rheum.; 39: 379-385.
42. Herrera-Esparza R, Barbosa-Cisneros O, Villalobos-Hurtado
26. Pelton BK, Hylton W, Denman AM. (1992). Activation of
R, Avalos-Diaz E. (1998). Renal expression of IL-6 and TNF
IL-6 production by UV irradiation of blood mononuclear cells
alpha genes in lupus nephritis. Lupus; 7: 154-158.
from patients with systemic lupus erythematosus. Clinical and
43. Takemura T, Yoshioka K, Murakami K, et al. (1994). Cellular
Experimental Immunology; 89(2): 251-254.
localization of inflammatory cytokines in human glomeru-
27. Klashman DJ, Martin RA, Martinez-Maza O, Stevens RH.
lonephritis. Virchows Arch.; 424: 459-464.
(1991). In vitro regulation of B cell differentiation by inter-
44. Hirohata S, Miyamoto T. (1990). Elevated levels of inter-
leukin-6 and soluble CD23 in systemic lupus erythematosus
leukin-6 in cerebrospinal fluid from patients with systemic
B cell subpopulations and antigen-induced normal B cells.
lupus erythematosus and central nervous system involvement.
Arthritis Rheum.; 34: 276-286.
Arthritis and Rheumatism; 33 (5): 644-649.
28. Swaak AJ, van der Brink HG, Aarden LA. (1996). Cytokine
45. Hirohata S, Hayakawa K. (1999). Enhanced interleukin-6
production (IL-6 and TNF alpha) in whole blood cell cultures
messenger RNA expression by neuronal cells in a patient with
of patients with systemic lupus erythematosus. Scand. J.
neuropsychiatric systemic lupus erythematosus. Arthritis and
Rheumatol.; 25: 233-238.
Rheumatism; 42 (12): 2729-2730.
29. Kitani A, Hara M, Hirose T, et al. (1992). Autostimulatory ef-
46. Hirohata S, Kanai Y, Mitsuo A, Tokano Y, Hashimoto H.
fects of IL-6 on excessive B cell differentiation in patients with
(2009). Accuracy of cerebrospinal fluid IL-6 testing for diag-
systemic lupus erythematosus: analysis of IL-6 production and
nosis of lupus psychosis. A multicenter retrospective study.
IL-6 expression. Clin. Exp. Immunol.; 88:75-83.
Clinical Rheumatology; 28 (11): 1319-1323.
30. Kitani A, Hara M, Hirose T, et al. (1989). Heterogeneity of
47. Esposito P, Balletta M.M. Procino A, Postiglione L, Memoli B.
B cell responsiveness to interleukin 4, interleukin 6 and low
(2009). Interleukin-6 release from peripheral mononuclear
molecular weight B cell growth factor in discrete stages of B
cells is associated to disease activity and treatment response
ISSN 1011 5528 www.smltsa.org.za 13
Medical Technology SA
Volume 25 No. 1 June2011
in patients with lupus nephritis. Lupus; 18(3): 1329-1330.
Quantitative polymerase chain reaction analysis reveals
48. De La Torre M, Urra JM, Blanco J. (2009). Raised expression
marked over expression of interleukin-1 beta, interleukin-1
of cytokine receptor gp130 subunit on peripheral lymphocytes
and interferon-gamma Mrna in the lymph nodes of lupus-
of patients with active lupus. A useful tool for monitoring the
prone mice. Molecular Immunology; 32: 495-503.
disease activity. Lupus; 18(3): 216-222.
63. Ishida H, Muchamuel T, Sakaguchi S, et al. (1994). Con-
49. IIIei GG, Shirota Y, Yarboro CH, et al. (2006). Tocilizumab
tinuous administration of anti-interleukin 10 antibodies delays
(humanized anti-IL6 Receptor Monoclonal Antibody) in pa-
onset of autoimmunity in NZB/W F1 mice. The Journal of
tients with systemic lupus erythematosus (SLE): safety, toler-
Experimental Medicine; 179: 305:310.
ability and preliminary efficacy. Arthritis and Rheumatism;
64. Llorente L, Richaud-Patin Y, Garcia-Padilla C, Claret E, Jakez-
54(12), supplement: p4043.
Ocampo J, Cardiel MH, Alcocer-Varela J, Grangeot-Kero L,
50. IIIei GG, Shirota Y, Yarboro CH, et al. (2010). Tocilizumab
Alarcón-Segovia D, Wijdenes J, Galanaud P, Emilie D. (2000).
in systemic lupus eryhtematosus: data on safety, preliminary
Clinical and biologic effects of anti-interleukin 10 monoclonal
efficacy and impact on circulating plasma cells from an open-
antibody administration in systemic lupus erythematosus. Ar-
label phase 1 dosage-escalation study. Arthritis and Rheuma-
thritis and Rheumatism; 43: 1790-1800.
tism; 62: 542-552.
65. Crispin JC, Liossis S-N C. Kis-Toth K, et al. (2010). Pathogen-
51. Ding L, Linsley S, Huang LY, Germain RN, Shevach EM.
esis of human systemic lupus erythematosus: recent advances.
(1993). IL-10 inhibits macrophage costimulatory activity by
Trends in Molecular Medicine; 16(2): 47-57.
selectively inhibiting the up-regulation of B7 expression. Jour-
66. Rouvier E, Luciani M-F, Mattei M-G, Denizot F, Golstein P.
nal of Immunology; 151(3): 1224-1234.
(1993). CTLA-8, cloned from an activated T cell, bearing AU-
52. De Wall Malefyt R, Haanen J, Spits H, et al. (1991). Inter-
rich messenger RNA instability sequences and homologous
leukin 10 (IL-10) and viral IL-10 strongly reduce antigen spe-
to a herpesvirus Saimiri gene. The Journal of Immunology;
cific human T cell proliferation by diminishing the antigen-
150(12): 5445-5456.
presenting capacity of monocytes via downregulation of class
67. Peck A, Mellins ED. (2010). Precarious balance: Th17 cells in
II major histocompatibility complex expression. Journal of
host defense. Infection and Immunity; 78(1):32-38.
Experimental Medicine; 174(4): 915-924.
68. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger
53. Fiorentiono DF, Bond MW, Mosmann TR. (1989). Two types
B. (2006). TGFb in the context of an inflammatory cytokine
of mouse T helper cell. IV. Th2 clones secrete a factor that
milieu supports de novo differentiation of IL-17 producing T
inhibits cytokine production of Th1 clones. Journal of Experi-
cells. Immunity; 24: 179-189.
mental Medicine; 170(6): 2081-2095.
69. Yang L, Anderson DE, Baaecher-Allan C, et al. (2008). IL-
54. Rousset F, Garcia E, Defrance T, et al. (1992). Interleukin 10
21 and TGF-b are required for differentiation of human TH17
is a potent growth and differentiation factor for activated hu-
cells. Nature; 454: 350-352.
man B lymphocytes. Proceedings of the National Academy of
70. Mangan PR, Harrington LE, O'Quinn DB, et al. (2006). Trans-
Sciences of the United States of America; 89(5): 1890-1893.
forming growth factor- b induces development of TH17 line-
55. López P, Gutiérrez C, Suárez A. (2010). IL-10 and TNFa
age. Nature; 441: 231-234.
genotypes in SLE. Journal of Biomedicine and Biotechnology;
71. Aggarwal S, Ghilardi N, Xie M-H, De Sauvage FJ, Gurney AL.
Volume 2010, Article ID 838390, 11 pages.
(2003). Interleukin-23 promotes a distinct CD4 T cell activa-
56. Van der Linden MW, Westendoep RG, Sturk A, et al. (2000).
tion state characterized by the production of interleukin-17.
High interleukin-10 production in first-degree relatives of pa-
Journal of Biological Chemistry; 278(3): 1910-1914.
tients with generalized but not cutaneous lupus erythemato-
72. Alabanesi C, Cavani A, Girolomoni G. (1999). IL-17 is pro-
sus. Journal of Investigative Medicine; 48: 327-334.
duced by nickel-specific T lymphocytes and regulates ICAM-1
57. Grodal G, Kristjansdottir H, Gunnlaugsdottir B, et al. (1999).
expression and chemokine production in human keratinoc-
Increased number of interleukin-10 producing cells in system-
ytes: synergistic or antagonistic effects with IFN-g and TNF-a.
ic lupus erythematosus patients and their first-degree relatives
The Journal of Immunology; 162(1): 494-502.
and spouses in Icelandic multicase families. Arthritis and
73. Nalbandian A, Crispin JC, Tsokos GC. (2009). Interleukin 17
Rheumatism; 42: 1649-1654.
and systemic lupus erythematosus: current concepts. Clinical
58. Alarcon-Riquelme ME, Lindqvist AK, Jonasson I, et al. (1999).
and Experimental Immunology; 157(2): 209-215.
Genetic analysis of the contribution of IL10 to systemic lupus
74. Crispin JC, Oukka M, Bayliss G, et al. (2008). Expanded dou-
erythematosus. The Journal of Rheumatology; 26: 2148-2152.
ble negative T cells in patients with systemic lupus erythema-
59. Ronnelid J, Tejde A, Mathsson L, et al. (2003). Immune com-
tosus produce IL-17 and infiltrate the kidneys. The Journal of
plexes from SLE sera induce IL-10 production from normal
Immunology; 181(12): 8761-8766.
peripheral blood mononuclear cells by a FcgammaRII de-
75. Wong CK, Lit LCW, Tam LS, Li EKM, Wong PTY, Lam CWK.
pendent mechanism: implications for a possible vicious cycle
(2008). Hyperproduction of IL-23 and IL-17 in patients with
maintaining B cell hyperactivity in SLE. Annals of Rheumatic
systemic lupus erythematosus: implications for Th17-medi-
Diseases; 62: 37-42.
ated inflammation in auto-immunity. Clinical Immunology;
60. Moulin V, Andris F, Thielemans K, et al. (2000). B lymphocytes
127(3): 385-393.
regulate dendritic cell (DC) function in vivo: increased inter-
76. Wong CK, Ho CY, Li EK, Lam CWK. (2000). Elevation of
leukin 12 production by DCs from B cell deficient mice results
proinflammatory cytokine (IL-18, IL-17, IL-12) and Th2 cy-
in T helper cell type 1 deviation. The Journal of Experimental
tokine (IL-4) concentrations in patients with systemic lupus
Medicine; 192: 475-482.
erythematosus. Lupus; 9(8): 589-593.
61. Tieng AT, Peeva E. (2008). B-cell directed therapies in sys-
77. Doreau A, Belot A, Bastid J, et al. (2009). Interleukin 17 acts
temic lupus erythematosus. Semin. Arthritis Rheum.; 38(3):
in synergy with B cell-activating factor to influence B cell bi-
ology and the pathophysiology of systemic lupus erythemato-
62. Prud'homme GJ, Kono DH, Theofilopoulos AN. (1995).
sus. Nature Immunology; 10(7): 778-785.
14 www.smltsa.org.za ISSN 1011 5528
Medical Technology SA
Volume 25 No. 1 June 2011
78. Crispin JC, Tsokos GC. (2009). Human TCR-ab+CD4-CD8- T
temic lupus erythematosus patients. Mod. Rheumatol.; 17:
cells can derive from CD8+ T cells and display an inflamma-
tory effector phenotype. The Journal of Immunology; 183(7):
97. Hillyer P, Larché MJ, Bowman EP, et al. (2009). Investigating
the role of the interleukin-23/-17A axis in rheumatoid arthritis.
79. Dong G, Ye R, Shi W, Liu S, Wang T, Yang X, Yang N, Yu X.
Rheumatology; 48: 1581-1589.
(2003). IL-17 induces autoantibody overproduction and
98. Kwan BC, Tam LS, Lai KB, et al. (2009). The gene expression
peripheral blood mononuclear cell over-expression of IL-6
of type 17 T-helper cell-related cytokines in the urinary sedi-
in lupus nephritis patients. Chinese Medical Journal (Engl);
ment of patients with systemic lupus erythematosus. Rheuma-
80. Ghilardi JC, Martinez A, Alcocer-Varela J. (2003). Quanti-
99. Chan RW, Lai FM, Li EK, et al. (2006). Imbalance of Th1/Th2
fication of regulatory T cells in patients with systemic lupus
transcription factors in patients with lupus nephritis. Rheuma-
erythematosus. J. Autoimmun; 21: 273-276.
tology; 45: 951-957.
81. Kikly K, Liu L, Na S, Sedgwick JD. (2006). The IL-23/Th17
100. Chan RW, Lai FM, Li EK, et al. (2007). Expression of T-bet,
axis: therapeutic targets for autoimmune inflammation. Curr.
a type 1 T-helper cell transcription factor, in the urinary sedi-
Opin. Immunol; 18: 670-675.
ment of lupus patients predicts disease flare. Rheumatology;
82. Graham KL, lee LY, Higgins JP, Steinman L, Utz PJ, Ho PP.
(2008). Failure of oral atorvastatin to modulate a murine mod-
101. Leng RX, Pan HF, Chen GM, Wang C, Qin WZ, Chen LL, Tao
el of systemic lupus erythematosus. Arthritis Rheum.; 58(7):
JH, Ye DQ. (2010). IL-23: A promising Therapeutic Target
for Systemic Lupus Erythematosus. Archives of Medical Re-
83. Lawman S, Mauri C, Jury EC, Cook HT, Ehrenstein MR. (2004).
Atorvastatin inhibits autoreactive B cell activation and delays
102. Schneider P, et al. (1999). BAFF, a novel ligand of the tumour
lupus development in New Zealand black/white F1 mice. J.
necrosis factor family, stimulates B cell growth. J. Exp. Med.;
Immunol; 173: 7641-7646.
189: 1747:1756.
84. Zhang X, Jin J, Peng X, Ramgolam VS, Markovic-Plese S.
103. Moore PA, et al. (1999). BLys: member of the tumour necro-
(2008). Simvastatin inhibits IL-17 secretion by targeting mul-
sis factor family and B lymphocyte stimulator. Science; 285:
tiple IL-17-regulatory cytokines and by inhibiting the expres-
sion of IL-17 transcription factor RORC in CD4-lymphocytes.
104. Shu HB, Johnson H. (2000). B cell maturation protein is a
J. Immunol.; 180: 6988-6996.
receptor for the tumour necrosis factor family member TALL-
85. Bianchi S, Bigazzi R, Caiazza A, Campese VM. (2003). A
1. Proc. Natl. Acad. Sci. U.S.A.; 97: 9156-9161.
controlled, prospective study of the effects of atorvastatin on
105. Mukhopadhyay A, Ni J, Zhai Y, Yu GL, Aggarwal BB. (1999).
proteinuria and progression of kidney disease. Am. J. Kidney
Identification and characterization of a novel cytokine,
Dis.; 41: 565-570.
THANK, a TNF homologue that activates apoptosis, nuclear
86. Cornelissen F, van Hamburg JP, Lubberts E. (2009). The IL-12/
factor-kappa B, and c-JunNH2-terminal kinase. J. Biol. Chem.;
IL-23 axis and its role in Th17 cell development, pathology
274: 15978-15981.
and plasticity in arthritis. Curr. Opin. Investig. Drugs; 10: 452-
106. Litinskiy MB, et al. (2002). DCs induce CD40-independent
immunoglobulin class switching through BLyS and APRIL.
87. Iwakura Y, Ishigame H. (2006). The IL-23/IL-17 axis in inflam-
Nat. Immunol.; 3: 822-829.
mation. J. Clin. Invest.; 116: 1218-1222.
107. Nardelli B, et al. (2001). Synthesis and release of B-lym-
88. Zhang Z, Kyttaris VC, Tsokos GC. (2009). The role of IL-23/
phocyte stimulator from myeloid cells. Blood; 97: 198-204.
IL-17 axis in lupus nephritis. J. Immunol.; 183: 3160-3169.
108. Lavie F, et al. (2008). Expression of BAFF(BLyS) in T cells
89. Langrish CL, McKenzie BS, de Waal Wilson NJ, et al. (2004).
infiltrating labial salivary glands from patients with Sjogren's
IL-12 and IL-23: master regulators of innate and adaptive im-
syndrome. J. Pathol.; 202: 496-502.
munity. Immunol. Rev.; 202: 96-105.
109. Scapini P, et al. (2003). G-CSF-stimulated neutrophils are
90. Kastelein RA, Hunter CA, Cua DJ. (2007). Discovery and bi-
a prominent source of functional BLyS. J. Exp. Med.; 197:
ology of IL-23 and IL-27: related but functionally distinct regu-
lators of inflammation. Annu. Rev. Immunol.; 25:221-242.
110. Ogden CA, et al. (2005). Enhanced apoptotic cell clearance
91. Pan HF, Ye DQ, Li XP. (2008). Type 17 T helper cells might be
capacity and B cell survival factor production by IL-10-acti-
a promising therapeutic target for systemic lupus erythemato-
vated macrophages: implications for Burkitt's lymphoma. J.
sus. Nat. Clin. Pract .Rheumatol.; 4: 352-353.
Immunol.; 174: 3015-3023.
92. Ooi JD, Phoon RK Holdsworth SR, et al. (2009). IL-23 and
111. Bossen C, Cachero A, Tardivel A, et al. (2008). TACI, unlike
not IL-12, directs autoimmunity to the Goodpasture antigen.
BAFF-R, is solely activated by oligomeric BAFF and APRIL to
J. Am. Soc. Nephrol.; 20: 980-989.
support survival of activated B cells and plasmablasts. Blood;
93. Mok CC, Lau CS, et al. (2003). Pathogenesis of systemic lu-
111(3): 1004-1012.
pus erythematosus. J. Clin. Pathol.; 56: 481-490.
112. Cheema GS, Roschke V, Hilbert DM, Stohl W. (2001). Elevat-
94. Yan Tan Z, Bealgey KW, Fang Y, Ming Gong Y, Bao S. (2008).
ed serum B lymphocyte stimulator levels in patients with sys-
Interleuken-23: Immunological roles and clinical implica-
temic immune-based rheumatic diseases. Arthritis & Rheum.;
tions. The International Journal of Biochemistry and Cell Biol-
44(6): 1313-1319.
ogy; 41: 733-735.
113. Hondowicz BD, et al. (2007). The role of BLyS/BLyS recep-
95. Duerr RH, Taylor KD, Brant SR, et al. (2006). A genome-wide
tors in anti-chromatin B cell regulation. Int. Immunol.; 19:
association study identifies IL23R as an inflammatory bowel
disease gene. Science.; 314: 1461-1496.
114. Cancro MP, D'Cruz DP, Khamashta MA. (2009). The role of
96. Huang XF, Hua J, Shen N, et al. (2007). Dysregulated ex-
B lymphocyte stimulator (BLyS) in systemic lupus erythemato-
pression of interleukin-23 and interleukin-12 subunits in sys-
sus. J. Clin. Invest.; 119: 1066:1073.
ISSN 1011 5528 www.smltsa.org.za 15
Medical Technology SA
Volume 25 No. 1 June2011
115. Petri M, Stohl W, Chatham W, et al. (2008). Association of
131. Lee HM, Mima T, Sugino H, et al. (2009). Interactions among
plasma B lymphocyte stimulator levels and disease activity in
type I and type II interferon, tumor necrosis factor, and b-es-
systemic lupus erythematosus. Arthritis Rheum.; 58: 2453-
tradiol in the regulation of immune response-related gene ex-
pressions in systemic lupus erythematosus. Arthritis Research
116. Stohl W, Metyas S, Tan S-M, et al. (2003). B lymphocyte
&Therapy; 11(1) article R1.
stimulator overexpression in patients with systemic lupus ery-
132. Bennett L, Palucka K, Arce E, et al. (2003). Interferon and
thematosus: longitudinal observations. Arthritis & Rheuma-
granulopoiesis signaturesin systemic lupus erythematosus
tism; 48(12): 3475-3486.
blood. Journal of Experimental Medicine; 197(6): 711-723.
117. Furie R, Stohl W, Ginzler EM, et al. (2008). Biologic activ-
133. Baechler EC, Batliwalla FM, Karypis G, et al. (2003). Inter-
ity and safety of belimumab, a neutralizing anti-B-lymphocyte
feron inducible gene expression signature in peripheral blood
stimulator (BLyS) monoclonal antibody: a phase I trial in pa-
cells of patients with severe lupus. Proceedings of the Na-
tients with systemic lupus erythematosus. Arthritis Research
tional Academy of Sciences of the United States of America;
and Therapy; 10(5): article R109.
100(5): 2610-2615.
118. Wallace DJ, Stohl W, Furie RA, et al. (2009). A phase II,
134. Han GM, Chen L, Shen N, Ye S, Bao CD, Gu YY. (2003).
randomized, double-blind, placebo controlled, dose-ranging
Analysis of gene expression profiles in human systemic lupus
study of belimumab in patients with active systemic lupus ery-
erythematosus using oligonucleotide microarray. Genes and
thematosus. Arthritis Rheum.; 61: 1168-1178.
Immunity; 4(3): 177-186.
119. Petri MA, Furie R, Merrill JT, et al. (Abstract 2009). Four year
135. Ishii T, Onda H, Tanigawa A, et al. (2005). Isolation and
experience of belimumab, a BLyS-specific inhibitor, in sys-
expression profiling of genes upregulated in the peripheral
temic lupus erythematosus (SLE). American College of Rheu-
blood cells of systemic lupus erythematosus. DNA Research;
matology National Meeting 2009.
12(6): 429-439.
120. GlaxoSmithKline and Human Genome Sciences announce
136. Feng X, Wu H, Grossman M, et al. (2006). Association of
topline 76-week results of phase 3 trial of Benlysta™ in sys-
increased interferon-inducible gene expression with disease
temic lupus erythematosus. Issued: Tuesday 20 April 2010,
activity and lupus nephritis in patients with systemic lupus
London UK & Rockville, Maryland US. www.gsk.com/media/
erythematosus. Arthritis and Rheumatism; 54(9): 2951-2962.
137. Bauer JW, Baechler EC, Petri M, et al. (2006). Elevated serum
January 3, 2011.
levels of interferon-regulated chemokines are biomarkers for
121. Al-Janadi M, Al-Balla S, Al-Dalaan, Raziuddin S. (1993). Cy-
active human systemic lupus erythematosus. PLoS Medicine;
tokine profile in systemic lupus erythematosus, rheumatoid
3(12) article e491: 2274-2284.
arthritis and other rheumatic diseases. Journal of Clinical Im-
138. Fu Q, Chen X, Cui H, et al. (2008). Association of elevated
munology; 13(1): 58-67.
transcript levels of interferon-inducible chemokines with dis-
122. Hooks JJ, Moutsopoulos HM, Notkins AL. (1981). Circulating
ease activity and organ damage in systemic lupus erythema-
interferon in human autoimmune diseases. Texas Reports on
tosus patients. Arthritis Research and Therapy; 10(5) article
Biology and Medicine; 41: 164-168.
123. Ytterberg SR, Schnitzer TJ. (1982). Serum interferon levels
139. Yao Y, Richman L, Higgs BW, et al. (2009). Neutralization
in patients with systemic lupus erythematosus. Arthritis and
of interferon-alpha/beta-inducible genes and downstream ef-
Rheumatism; 25(4): 401-406.
fect in a phase I trial of an anti-interferon-alpha monoclonal
124. Kim T, Kanayama N, Negoro N, Okamura M, Takeda T, Inoue
antibody in systemic lupus erythematosus. Arthritis Rheum.;
T. (1987). Serum levels of interferons in patients with sys-
60: 1785-1796.
temic lupus erythematosus. Clinical and Experimental Immu-
140. Clinical Trials.gov. A study to evaluate safety and tolerability
nology; 70(3): 562-569.
of IV or SC dose of MEDI-545 in patients with systemic lupus
125. Robak E, Smolewski P, Wozniacka A Sysa-Jedrzejowska A,
Stepien H, Robak T. (2004). Relationship between periph-
show/NCT01031836 Accessed January 5, 2011.
eral blood dendritic cells and cytokines involved in the patho-
141. Clinical Trials.gov. A study to evaluate safety and toler-
genesis of systemic lupus erythematosus. European Cytokine
ability of subcutaneous doses of MEDI-545 in subjects with
Network; 15(3): 222-230.
126. Bengtsson AA, Sturfelt G, Truedsson L, et al. (2000). Activa-
NCT00657189 Accessed January 5, 2011.
tion of type I interferon system in systemic lupus erythemato-
142. Theofilopoulos AN, Lawson BR. (1999). Tumour necrosis fac-
sus correlates with disease activity but not with antiretroviral
tor and other cytokines in murine lupus. Annals of the Rheu-
antibodies. Lupus; 9(9): 664-671.
matic Diseases; 58(supplement 1): 149-155.
127. Dall'era MC, Cardarelli PM, Preston BT, Witte A, Davis Jr JC.
143. Aringer M, Smolen JS. (2003). Complex cytokine effects in
(2005). Type I interferon correlates with serological and clini-
a complex autoimmune disease: tumor necrosis factor in sys-
cal manifestations of SLE. Annals of the Rheumatic Diseases;
temic lupus erythematosus. Arthritis Research and Therapy;
64(12): 1692-1697.
128. Wenzel J, Zahn S, Bieber T, Tüting T. (2009). Type I inter-
144. Gabay C, Cakir N, Moral F, et al. (1997). Circulating levels
feron-associated cytotoxic inflammation in cutaneous lupus
of tumor necrosis factor soluble receptors in systemic lupus
erythematosus. Archives of Dermatological Research; 301(1):
erythematosus are significantly higher than in other rheumatic
disease and correlate with disease activity. Journal of Rheu-
129. Takeuchi O, Akira S. (2009). Innate immunity to virus infec-
matology; 24(2): 303-308.
tion. Immunological Reviews; 227(1): 75-86.
145. Aringer M, Smolen JS. (2004). TNF and other proinflamma-
130. Fitzgerald-Bocarsly P, Dai J, Singh S. (2008). Plasmacytoid
tory cytokines in SLE: A rationale for therapeutic intervention.
dendritic cells and type I IFN: 50 years of convergent history.
Lupus; 13: 344-347.
Cytokine and Growth Factor Reviews; 19(1): 3-19.
146. Aringer M, Smolen JS. (2005). Cytokine expression in lupus
16 www.smltsa.org.za ISSN 1011 5528
Medical Technology SA
Volume 25 No. 1 June 2011
kidneys. Lupus; 14: 189-191.
events and efficacy of TNF-alpha blockade with infliximab in
147. Mohan AK, Edwards ET, Cote TR, Siegal JN, Braun M. (2002).
patients with systemic lupus erythematosus: long term follow-
Drug-induced systemic lupus erythematosus and TNF-a
up of 13 patients. Rheumatology (Oxford); 48: 1451-1454.
blockers. The Lancet; 360(9333): 646.
153. Matsumura R, Umemiya K, Sugiyama T, et al. (2009). Anti-
148. Shakoor N, Michalska M, Harris CA, Block JA. (2002). Drug-
tumor necrosis factor therapy in patients with difficult-to-treat
induced systemic lupus erythematosus associated with etaner-
lupus nephritis: a prospective series of nine patients. Clin.
cept therapy. The Lancet; 359(9306): 579-580.
Exp. Rheumatol.; 27: 416-421.
149. Neale TJ, Ruger BM, Macaulay H, et al. (1995). Tumor necro-
154. Takahashi N, Naniwa T, Banno S. (2008). Successful use of
sis factor-a is expressed by glomerular visceral epithelial cells
etanercept in the treatment of acute lupus hemophagocytic
in human membranous nephropathy. American Journal of Pa-
syndrome. Mod. Rheumatol.; 18:72-75.
thology; 146(6): 1444-1454.
155. Uppal SS, Hayat SJ, Raghupathy R. (2009). Efficacy and safety
150. Pisetsky DS. (2000). Tumor necrosis factor alpha blockers
of infliximab in active SLE: a pilot study. Lupus; 18:690-697.
and the induction of anti-DNA autoantibodies. Arthritis and
156. Clinical Trials.gov. TNF blockade with remcade in active lu-
Rheumatism; 43: 2381-2382.
pus nephritis WHO class V (TRIAL) (NCT00368264) http://
151. Aringer M, Steiner G, Graninger W, et al. (2001). Role of
clinicaltrials.gov/ct/show/NCT00368264 Accessed January 6,
tumor necrosis factor alpha and potential benefit of tumor
necrosis factor blockade treatment in systemic lupus ery-
157. Clinical Trials.gov. Etanercept for the treatment of lupus ne-
thematosus: comment on the editorial by Pisetsky. Arthritis
and Rheumatism; 44: 1721-1722.
NCT00447265 Accessed January 6, 2011.
152. Aringer M, Houssiau F, Gordon C, et al. (2009). Adverse
Peer reviewed original articlE
INTERACTION BETWEEN NK CELLS AND HLA-G1 AT THE PLACENTAL
INTERFACE OF HIV-1 INFECTED PREGNANT WOMEN: ADDITIONAL
RISK FACTORS OR PHYSIOLOGICAL ASSOCIATION?
s Moodley (phd)
Department of Biomedical Sciences, Mangosuthu University of Technology, South Africa
Grants: Support for the study was received from The National Research Funding- Thuthuka Programme and Mangosuthu University of
Technology Research Grant.
Corresponding author: Shamala Moodley email: [email protected] tel: +27 (0)31 907 7450 fax: +27 (0)31 907 7451
ABSTRACTBackground: Human Leucocyte Antigen-G (HLA-G) molecules are involved in the inhibition of cell-mediated immune responses and could promote the propagation of HIV-1 infection across the placental interface thus increasing the risk of vertical transmission. Therefore, the objective of this study was to assess whether the Major Histocompatibility Complex (MHC) - coded molecule HLA-G inhibits Natural Killer (NK) cell activity thereby, assisting viral penetration across the placental barrier in HIV-1 positive pregnant women.
Study Design & Methods: Natural Killer (CD56+) cell activity and placental HLA-G1 expression was assessed using immunohisto-chemistry and real-time polymerase chain reaction (RT-PCR) techniques, respectively. Studies were performed on a total of fifty five placental samples obtained from HIV-1 infected mothers at birth. Results: Low numbers of NK cells increased risk of vertical transmission [OR = 3.424 (95%CI 0.65-17.89)]. The risk of babies becoming infected increased by 1.3 with every 1 unit increase in HLA-G1 expression. A positive correlation was observed between mothers' log viral load and transmission of infection to the baby (p = 0.047; 95%CI 1.029-11.499). Conclusion: Low NK cell activity at the placental interface increased the risk of vertical transmission. Maternal viral load remained a strong predictor of viral transmission.
KEYWORDSNatural Killer cells (CD56+), Human Leucocyte Antigen-G1, vertical transmission, viral load, up regulation.
Natural Killer (NK) cells are a population of low-density, large
against certain microbial, viral and parasitic infections [1, 2]. In
granular lymphocytes, which mainly develop and differenti-
response to proinflammatory stimuli, which may be induced by
ate in bone marrow and then enter into the circulation. These
a viral infection, NK cells migrate to various tissues and organs
cells comprise approximately 5-20% of peripheral blood
of the body. In the mucosal decidual tissues of the maternal
lymphocytes and are involved in the innate immune response
uterus, NK cells are the most abundant class of lymphocyte,
ISSN 1011 5528 www.smltsa.org.za 17
Source: http://mtsaj.co.za/index.php/mtsaj/article/viewFile/14/13
Submitted By: Parish Disaster Committees Date of Event: August 28, 2008 St. James Type: Tropical Storm Gustav INTRODUCTION AND BACKGROUND Summary:Tangle River - (break away of a section of the road to the bottom end and upper middle a long crack in the surface. Johnson - huge landslide blocking more than half the roadway bringing traffic to a snarl. Further into Johnson before reaching the Church there was another huge breakaway in the road. Heading to Niagra/Arcadia were flooded waters across the main roads.
La combinación de fármacos en la artrosis: una realidad terapéutica. EDITORIAL LITERATURA INTERNACIONAL Ctra. Nacional II. km 680,608389 Palafolls (Barcelona) – EspañaTel. 93 490 49 08 Fax 93 490 97 11 • Eficacia de la combinación de hidrocloruro de glucosamina, condroitín sulfato sódico de bajo peso molecular y ascorbato de manganeso en el tratamiento de la artrosis de rodilla