Kamagra gibt es auch als Kautabletten, die sich schneller auflösen als normale Pillen. Manche Patienten empfinden das als angenehmer. Wer sich informieren will, findet Hinweise unter kamagra kautabletten.
Oxidative stress as pathogenesis of cardiovascular risk associated with metabolic syndrome
ANTIOXIDANTS & REDOX SIGNALINGVolume 15, Number 7, 2011
FORUM REVIEW ARTICLE
ª Mary Ann Liebert, Inc.
DOI: 10.1089/ars.2010.3739
Oxidative Stress as Pathogenesis
of Cardiovascular Risk Associated with Metabolic Syndrome
Metabolic syndrome (MetS) is characterized by accumulation of visceral fat associated with the clustering ofmetabolic and pathophysiological cardiovascular risk factors: impaired glucose tolerance, dyslipidemia, andhypertension. Although the definition of MetS is different among countries, visceral obesity is an indispensablecomponent of MetS. A growing body of evidence suggests that increased oxidative stress to adipocytes is centralto the pathogenesis of cardiovascular disease in MetS. Increased oxidative stress to adipocytes causes dysre-gulated expression of inflammation-related adipocytokines in MetS, which contributes to obesity-associatedvasculopathy and cardiovascular risk primarily through endothelial dysfunction. The purpose of present reviewis to unravel the mechanistic link between oxidative stress and cardiovascular risk in MetS, focusing on insulinresistance, hypertension, and atherosclerosis. Then, therapeutic opportunities translated from the bench tobedside will be provided to develop novel strategies to cardiovascular risk factors in MetS. Antioxid. RedoxSignal. 15, 1911–1926.
inflammatory state, which contributes to obesity-associatedvasculopathy and cardiovascular risk (12, 76, 87). These
Metabolic syndrome (MetS) is characterized by accu- adipocytokines are generally divided into pro-inflammatory
mulation of visceral fat associated with the clustering of
cytokines such as tumor necrosis factor-a, interleukin-6,
metabolic and pathophysiological cardiovascular risk factors:
monocyte chemoattractant protein-1, plasminogen activator
impaired glucose tolerance (IGT), dyslipidemia, and hyper-
inhibitor-1, and anti-inflammatory cytokines such as adipo-
tension (HTN) (47). Although the definition of MetS is dif-
nectin. Imbalance between pro-inflammatory cytokines and
ferent among countries, visceral obesity is an indispensable
anti-inflammatory cytokines is responsible for oxidative stress
component of MetS. The prevalence of MetS is rapidly in-
especially to endothelial cells and underlies the pathogene-
creasing worldwide not only in industrialized countries but
sis of the obesity-associated insulin resistance, IGT, type-2
also in developing countries associated with an increase in
diabetes mellitus (T2DM), HTN, dyslipidemia, and vascular
food intake. MetS has a strong impact on the global incidence
disease. Although obstructive sleep apnea syndrome repre-
of the life-threatening cardiovascular disease such as stroke
sents another important cause of oxidative stress in MetS
and myocardial infarction (2, 53). Although the MetS is mul-
(67, 77), this topic will not be discussed in this review be-
tifactorial in origin, IGT, dyslipidemia, and HTN are caused
cause unlike the increase in visceral adipose tissue, which
by the same underlying mechanism—endothelial dysfunction
is involved in a definition of MetS, not all the individuals
primarily mediated by oxidative stress.
with MetS are associated with obstructive sleep apnea
It is now apparent that visceral adipose tissue is an endo-
crine organ that secretes many bioactive molecules, known
The purpose of the present review is to overview the
as adipocytokines (20, 134, 152). The production of adipocy-
mechanistic link between oxidative stress and cardiovascular
tokines is of particular interest, because their local secretion
risk in MetS based on the evidence obtained from animal
by perivascular adipose depots may provide a new mecha-
experiments and clinical trials. This review specifically fo-
nistic link between obesity and its associated cardiovascu-
cusses on insulin resistance and atherosclerosis, which are
lar complications. Increased oxidative stress to adipocytes
intimately related to oxidative stress to endothelial cells. Then,
causes dysregulated expression of inflammation-related
therapeutic opportunities translated from the bench to bed-
adipocytokines in MetS. Increasing evidence supports the
side will be provided to develop novel strategies for pre-
central role of adipose tissue in the development of systemic
venting cardiovascular risk associated with MetS.
Second Department of Internal Medicine, Kansai Medical University, Moriguchi City, Japan.
Mechanisms Underlying Cardiovascular Risk in MetS
Central role of oxidative stressin visceral adipose tissue
A growing body of evidence suggests that increased oxi-
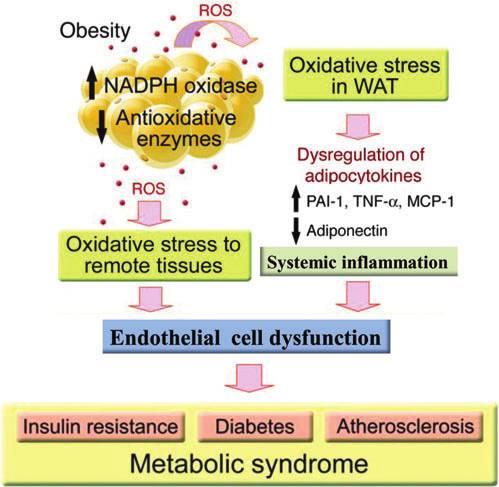
dative stress in white adipose tissue is central to the patho-genesis of cardiovascular disease in MetS. Although themolecular mechanism of oxidative stress to adipocytes re-mains unclear and appears to be multifactorial, the develop-ment of adipocyte hypertrophy and hypoxia has beenimplicated in oxidative stress (54). Reactive oxygen species(ROS) production increases in parallel with fat accumulationin adipocytes and increased levels of fatty acid stimulate ROSproduction in adipocytes through the activation of NADPHoxidase and decreased expression of antioxidative enzymes(49). Exposure of adipocytes to oxidative stress decreasesanti-inflammatory adiponectin (57, 133) and increases pro-inflammatory adipocytokines (23, 49, 121). Involvement of alocal renin-angiotensin aldosterone system (RAAS) has alsobeen proposed as a potential mediator of oxidative stress toadipocytes (22). Irrespective of the mechanism of ROS pro-duction, oxidative stress in the visceral adipose tissue is anupstream event that mediates systemic inflammation and
Mechanism of obesity-induced cardiovascular
oxidative stress in the remote tissue through dysregulation of
risk. In white adipose tissue (WAT) reactive oxygen species
adipocytokine production. Systemic inflammation then cau-
(ROS) production increases in parallel with fat accumulation
ses a variety of metabolic and cardiovascular disorders
in adipocytes through the activation of NADPH oxidase and
through oxidative stress to endothelial cells (Fig. 1).
decreased expression of antioxidative enzymes. Oxidativestress in WAT causes dysregulation of adipocytokines; in-creased generation of pro-inflammatory cytokines such as
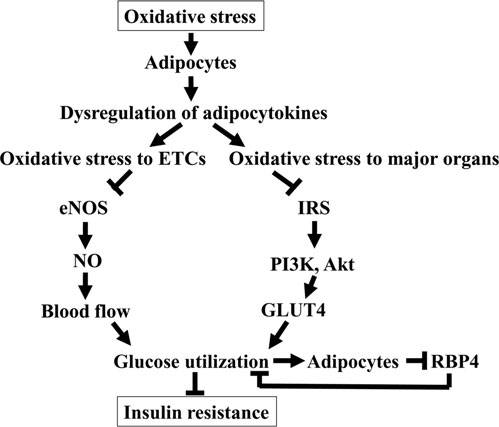
Oxidative stress and insulin resistance
plasminogen activator inhibitor (PAI)-1, tumor necrosis fac-tor-a (TNF-a), and monocyte chemoattractant protein-1
Oxidative stress to endothelial cells and subsequent de-
(MCP-1); and decreased generation of anti-inflammatory
crease in glucose uptake and utilization by major energy-
cytokines such as adiponectin. Dysregulation of adipocyto-
consuming organs such as the liver and skeletal muscle are
kines causes oxidative stress to remote tissues and systemic
responsible for insulin resistance. In MetS, endothelial cells
inflammation responsible for endothelial cell dysfunction,
are directly exposed to ROS through high levels of circulating
which is central to the pathogenesis of insulin resistance,
pro-inflammatory cytokines generated in the visceral adipose
diabetes, and atherosclerosis in metabolic syndrome (MetS).
tissue and low levels of adiponectin. Moreover, endothelial
This illustration is adapted from Furukawa et al. (49).
cell generation of ROS is increased by activation of NADPHoxidase through the action of local RAAS (31, 73). Indeed,RAAS-associated signaling by way of the angiotensin (Ang) II
metabolically active tissues contributing to the impairment of
type-1 receptors and mineral corticoid receptors triggers tis-
insulin-stimulated glucose and lipid metabolism.
sue activation of the NADPH oxidase and increased produc-
Another critical effect of ROS on the glucose uptake
tion of ROS in endothelial cells (132). This vicious cycle of ROS
mechanism is the activation of serine/threonine kinase
generation in endothelial cells is an important mechanism of
cascades such as c-Jun N-terminal kinase and nuclear factor-
transition from insulin resistance and IGT to T2DM in MetS.
kappaB, and others that in turn phosphorylate multiple
Oxidative stress to endothelial cells decreases bioavail-
targets, including the insulin receptor and the insulin receptor
ability of nitric oxide (NO) and causes loss of blood flow
substrate (IRS) proteins (14, 43, 104). Increased serine phos-
regulation in response to increased oxygen demand and en-
phorylation of IRS reduces its ability to undergo tyrosine
ergy utilization. Reduced bioavailability of NO results from
phosphorylation and may accelerate the degradation of IRS-1
decreased synthesis by uncoupling of endothelial NO syn-
(7), leading to the disruption of signaling pathways for glu-
thase (eNOS) through ROS-induced oxidation and depletion
cose uptake by glucose transporter-4 (GLUT4) through IRS-1
of the eNOS cofactor, tetrahydrobiopterin (BH4) (11, 125), in
and phosphatidylinositol 3-kinase (PI3K)/Akt. GLUT4 causes
combination with enhanced consumption in tissues by high
impairment of insulin-stimulated skeletal muscle glycogen
levels of superoxide generating peroxynitrite. This molecule is
synthesis, which appears to underlie the mechanism of insulin
highly toxic and causes endothelial cell death (36) that further
resistance (19).
reduces endothelial cell generation of NO. eNOS-derived NO
Impaired glucose uptake by adipocytes thorough the IRS-1,
also plays a crucial role in angiogenesis by upregulating
PI3K/Akt, and GLUT4 axis may cause an additional adverse
vascular endothelial growth factors and increasing mobiliza-
effect on insulin resistance. Adipose tissue has been proposed
tion of endothelial progenitor cells from the bone marrow (41,
to act as a glucose sensor (138). Adipocytes, therefore, detect
82). Thus, ROS-induced endothelial dysfunction impairs
the absence of glucose uptake by GLUT4 and, in response,
blood flow regulation and reduces expansion of the capillary
secrete adipocytokines such as retinol-binding protein 4 to
network, with attenuation of microcirculatory blood flow in
restrict glucose uptake in the skeletal muscle and increase
OXIDATIVE STRESS AND METABOLIC SYNDROME
glucose output by the liver by blocking insulin signaling (95),
models of HTN, a unifying characteristic of these models is
thereby increasing the blood glucose level. It was found that
the presence of oxidative stress that participates in the main-
the expression of GLUT4 is reduced in adipocytes, but not in
tenance of elevated arterial pressure and seems to be a com-
skeletal muscle, of animals and humans with obesity and
mon denominator underlying endothelial dysfunction in
T2DM (127). Thus, oxidative stress-induced downregulation
various forms of experimental HTN. In the presence of oxi-
of GLUT4 in adipocytes is a representative mechanism of in-
dative stress, eNOS acts as a double-edged sword. Superoxide
sulin resistance and T2DM in MetS. The putative model of
produced by inflammatory cells or endothelial cells stimu-
insulin resistance and T2DM in MetS is illustrated in Figure 2.
lated with pro-inflammatory adipocytokines react with NO,thereby stimulating the production of peroxynitrite. Perox-
Oxidative stress and HTN
ynitrite in turn causes uncoupling of eNOS, thereforeswitching an antiatherosclerotic NO-producing enzyme to
Endothelial dysfunction contributes to HTN, one of the
an enzyme that may accelerate the atherosclerotic process
diagnostic criteria of MetS. Reduced bioavailability of NO
by producing superoxide (69, 94). Besides circulating in-
appears to be a key process through which endothelial dys-
flammatory cells and pro-inflammatory adipocytokines, there
function is manifested in HTN. Accumulating evidence sug-
are a variety of sources of ROS in the vascular tissue. ROS-
gests that NO plays a major role in regulating blood pressure
producing enzymes involved in increased oxidative stress
and that impaired NO bioactivity is an important mechanism
within the vascular tissue include NADPH oxidase, xanthine
of HTN (78, 125, 141, 145). Mice with disruption of the gene
oxidase, and mitochondrial superoxide-producing enzymes.
for eNOS have elevated blood pressure levels compared with
Of these, local RAAS-mediated NADPH oxidase activation is
control animals (128, 135), suggesting a genetic component to
of prime importance in endothelial cell generation of ROS,
the link between impaired NO bioactivity and HTN. Al-
which contributes to endothelial dysfunction and HTN (122).
though the contribution of NO may vary between different
Oxidative stress-induced uncoupling of NOS is not con-
fined to eNOS. Oxidative stress on endothelial cells increasesexpression of inducible NOS (iNOS). Unlike eNOS, iNOS isconstitutively active and generates robust NO. Because oxi-dative stress depletes BH4 and uncouples iNOS, it is possiblethat iNOS uncoupling exaggerates oxidative stress and cre-ates a vicious cycle of endothelial dysfunction and HTN.
The potential role of NOS uncoupling in HTN and the ther-apeutic opportunity that targets NOS uncoupling will bediscussed later.
Oxidative stress and atherosclerosis
Atherosclerosis is one of the representative manifestations
of vascular pathology in MetS. Development of insulin resis-tance, HTN, and dyslipidemia culminates in atherosclerosis.
A growing body of evidence indicates that pro-inflammatorycytokines generated in the visceral adipose tissues are associ-ated with atherosclerosis. A sequence of events that are par-ticipated in the development of atheromatous plaque isillustrated in Figure 3.
One of the triggers of atheromatous plaque formation is
endothelial generation of ROS (Fig. 3A). Another initial par-
Schematic drawing of the role of oxidative stress
ticipant in atheromatous lesion-prone sites includes the inti-
to adipocytes in insulin resistance. Oxidative stress to adi-
mal influx and accumulation of low-density lipoprotein
pocytes causes dysregulation of adipocytokines that mediate
(LDL), which is further enhanced in the presence of triglyc-
systemic inflammation and oxidative stress to endothelial
eride. LDL is oxidized by ROS, and oxidized LDL is taken up
cells (ETCs) and major energy-consuming organs such as
by macrophages via their scavenger receptors CD36 to form
liver and skeletal muscles. Oxidative stress to ETCs promotes
foam cells (26, 115, 143). Monocyte-macrophage recruitment
uncoupling of endothelial nitric oxide synthase (eNOS) andreduces bioavailability of NO that impairs blood flow in
to the intima is likely to be regulated not only by a multiplicity
metabolically active tissues, leading to the impairment of
of adhesion molecules, integrins, and selectins, but also by
insulin-stimulated glucose utilization. Oxidative stress to
chemokines such as monocyte chemoattractant protein-1,
major energy-consuming organs, on the other hand, in-
which is constitutively synthesized and secreted by endo-
creases serine phosphorylation and degradation of insulin
thelial cells and smooth muscle cells (SMCs) migrated from
receptor substrate (IRS) and disrupts signaling pathways for
the media and adventitia (16, 153). Transcriptional upregu-
glucose uptake by glucose transporter-4 (GLUT4) through
lation of these molecules is enhanced by ROS, which are de-
the IRS-1 and phosphatidylinositol 3-kinase/Akt signaling
rived from endothelial cells, activated macrophages, and
pathway. Impaired glucose uptake by adipocytes thorough
SMCs. On the other hand, such ROS are also pivotal in the
GLUT4 causes secretion of retinol-binding protein 4 (RBP4)
oxidation of LDL, creating a self-perpetuating cycle in foam
to restrict glucose uptake in skeletal muscle and increaseglucose output by the liver, thereby reducing glucose utili-
cell accumulation and atherosclerotic plaque formation. At
zation and contributing to insulin resistance.
the same time, SMCs migrate from the media to the intimal
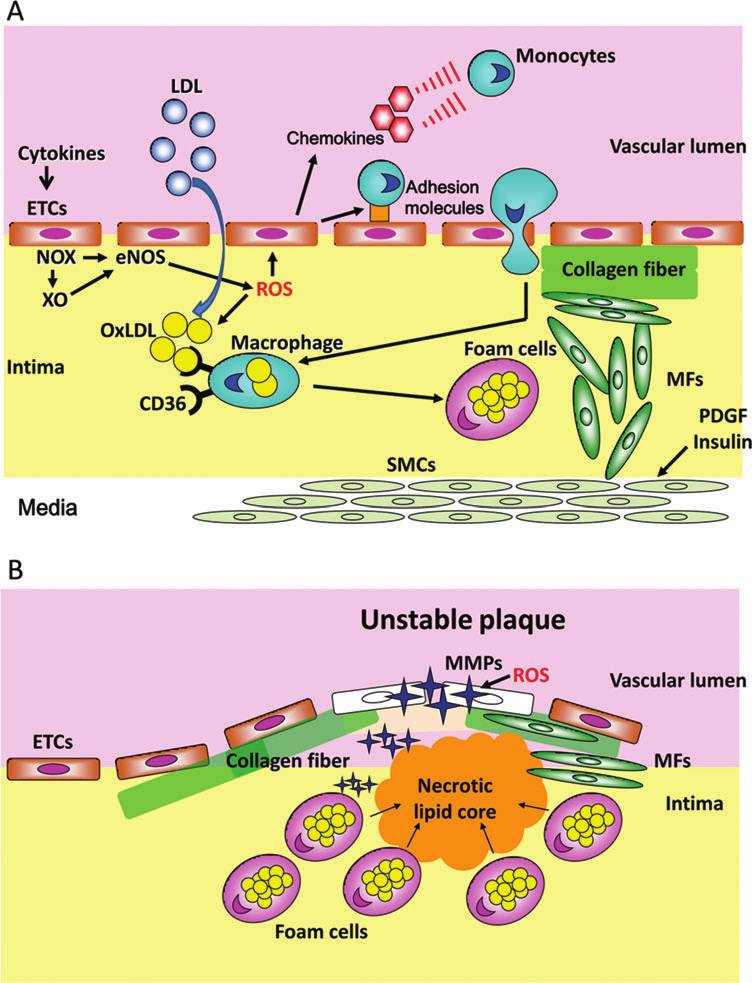
events participated in thedevelopment
tous plaque. (A) An earlyphase of atheromatous plaqueformation.
cytokines trigger endothelialcell (ETC) generation of ROSby activating NADPH oxidase(NOX) and xanthine oxidase(XO). These ROS promoteeNOS uncoupling and accen-tuate ROS generation. The in-timal influx and accumulationof
(LDL) represents another trig-ger of atheromatous plaqueformation. LDL is oxidizedby ROS, and oxidized LDL(OxLDL) is taken up by mac-rophages via their scavengerreceptors CD36 to form foamcells. ROS stimulate mono-cyte-macrophage recruitmentto the intima by enhanced ex-pression of adhesion mole-cules and chemokines. At thesame time smooth muscle cells(SMCs) migrate from the me-dia to the intimal endotheliallayer, differentiated into myo-fibroblasts (MFs), and prolif-erate under the regulation of anumber of mitogens, includ-ing platelet-derived growthfactor (PDGF) and insulin.
Proliferating MFs synthesizecollagen and promote thick-ening of the intima. (B) Alate stage of atheromatous pla-que development. A robust in-crease in inflammation andoxidative stress causes apopto-sis of foam cells, leading to theformation of the necrotic lipidcore. The necrotic lipid core iscovered with a thin fibrous cupcreating unstable plaque as aresult of degradation of colla-gen fiber by metalloproteinases(MMPs) in the presence of ROS.
endothelial layer. They are differentiated into myofibroblasts
trix metalloproteinases in the presence of ROS (79, 151). The
and proliferate under the regulation of a number of mitogens,
thin fibrous cup is prone to be ruptured in response to in-
including platelet-derived growth factor (37, 97). Insulin also
creased shear stress caused by elevated intraluminal pressure
acts as a growth factor and enhances intimal myofibroblast
and luminal narrowing of the coronary artery (92, 157).
growth when serine residues of IRS-1 are phosphorylated by
Therefore, such an advanced atheromatous plaque consisted
oxidative stress (65, 149). In addition, collagen synthesis by
of necrotic lipid core covered with a thin fibrous cap is termed
proliferating myofibroblasts is substantial for the thickening
unstable plaque.
of the intima.
In the late stage of plaque development (Fig. 3B), a robust
increase in inflammation and oxidative stress causes apo-
for Cardiovascular Risk Factors in MetS
ptosis of foam cells and is responsible for the formation of thenecrotic lipid core (64, 84). The necrotic lipid core is covered
Therapeutic approaches to MetS comprises lifestyle modi-
with a fibrous cup enriched with collagen fiber. However, the
fication in conjunction with drug treatment of the MetS-
fibrous cap is degraded by redox-sensitive activation of ma-
associated complications. Healthier eating and regular
OXIDATIVE STRESS AND METABOLIC SYNDROME
exercise greatly reduce waistline and body mass index, lower
described before. Although many model organisms have
blood pressure, and improve lipid profile. Lifestyle modifi-
consistently demonstrated positive responses to CR, it re-
cation has been shown to prevent T2DM development.
mains to be shown whether CR will extend lifespan in hu-
Nevertheless, appropriate treatment of cardiovascular risk
mans. The first results from a long-term, randomized,
factors in MetS often requires pharmacologic intervention
controlled CR study in nonhuman primates showing statis-
against IGT or T2DM with insulin-sensitizing agents, such as
tically significant benefits on longevity have now been re-
thiazolidinediones (TDZs) and metformin, whereas statins
ported (27). Additionally, positive results from short-term,
and fibrates or angiotensin-converting enzyme (ACE) in-
randomized, controlled CR studies in humans are suggestive
hibitors and Ang II type-1 receptor blockers (ARBs) are the
of potential health and longevity gains (61). However, the
first-line lipid-modifying or anti-HTN drugs. These pharma-
current environment of excess caloric consumption and high
cological interventions inhibit oxidative stress, but unlike
incidence of overweight/obesity illustrate the improbable
general antioxidants of which efficacy to prevent cardiovas-
nature of the long-term adoption of a CR lifestyle by a sig-
cular risk factors is still controversial, they prevent only
nificant proportion of the human population. Thus, the search
harmful ROS generation, leaving beneficial ROS. Thus, these
for substances that can reproduce the beneficial physiologic
drugs are designated as a class of preventive antioxidants en-
responses of CR without a requisite calorie intake reduction,
abling a causal therapy against oxidative stress through site-
termed CR mimetics, has gained momentum.
specific inhibition of ROS and preservation of redox signaling
The molecular mechanism underlying the efficacy of re-
necessary for cardiovascular protection (107). Further, pre-
duction of visceral fat mass by CR to reduce cardiovascular
ventive antioxidants appear to increase eNOS-derived NO,
risk factors may be related to increased generation of adipo-
which prevents insulin resistance, HTN, and atherosclerosis
nectin in the visceral adipocytes (158). It has been demon-
(98). Discussed in this section are therapeutic opportunities for
strated that CR in rats significantly increases the level of
cardiovascular risk factors in MetS focusing on the strategy to
circulating adiponectin, a distinctive marker of differentiated
inhibit oxidative stress and inflammation in the visceral adi-
adipocytes (167). PPAR-c is a member in the nuclear receptor
pose tissue and preserve endothelial functions NO generation
superfamily that mediates part of the regulatory effects of
that are central to prevent cardiovascular risk factors in MetS.
dietary fatty acids on gene expression and may be a molecularlink between CR and increased generation of adiponectin. CRfor 2 and 25 months, significantly increased the expression of
Caloric restriction and adiponectin
PPAR-c, C/EBPb, and Cdk-4, and partially attenuated age-
A large body of experimental and epidemiological evi-
related decline in C/EBPa expression relative to rats fed ad
dence has established an association between visceral obesity
libitum (166). As a result, adiponectin was upregulated at both
and MetS. Caloric restriction (CR) primary affects energy
mRNA and protein levels, resulting in activation of target
stores in visceral adipose tissue (32). Indeed, a substantial
genes involved in fatty acid oxidation and fatty acid synthesis.
improvement in all aspects of MetS with only a moderate
Moreover, CR significantly decreased the ratio of C/EBPb
degree of weight loss by CR has been observed in a large
isoforms LAP/LIP, suggesting the suppression of gene tran-
number of randomized, controlled studies and can also be
scription associated with terminal differentiation while facil-
obtained in severe obesity, despite the fact that the patients
itating preadipocytes proliferation. Morphometric analysis
remain obese (33). The reasons for this apparent dissociation
revealed a greater number of small adipocytes in CR relative
between weight loss and metabolic improvement are not yet
to ad libitum feeding. Immunostaining confirmed that small
clearly understood, but may involve the relationship between
adipocytes were more strongly positive for adiponectin than
visceral fat and metabolic alterations. The results of some
the large ones. Overall, these results suggest that CR increased
studies suggest that the favorable metabolic changes observed
the expression of adipogenic factors and maintained the dif-
in obese patients with CR and weight loss may be directly
ferentiated state of adipocytes, which is critically important
attributable to a reduction in visceral fat (59). Moreover, vis-
for adiponectin biosynthesis. On the other hand, adiponectin
ceral adipose tissue is a pivotal organ in aging process and in
is a CR mimetic. It has been demonstrated that mice with
the determination of life span. There is growing evidence that
transgenic expression of human adiponectin that had persis-
the effect of reduced adipose tissue mass on life span could be
tent hyperadiponectinemia exhibited significantly decreased
due to the prevention of obesity-related metabolic disorders,
weight gain associated with less fat accumulation and smaller
including T2DM and atherosclerosis (15).
adipocytes in both visceral and subcutaneous adipose tis-
The mechanism underlying improvements of the aspect of
sues (106). Macrophage infiltration in adipose tissue was
MetS and prevention of aging by CR has been extensively
markedly suppressed in the transgenic mice. In the hyper-
investigated. Aging is associated with increased visceral fat,
adiponectinemic mice, daily food intake was not altered, but
and recent studies suggested that visceral fat could influence
oxygen consumption was significantly greater, suggesting
longevity (88, 96, 163). It has recently been proposed that si-
increased energy expenditure. Moreover, high-calorie diet–
lent information regulator 2 (SIR2) ortholog, sirtuin 1 (SIRT1),
induced premature death was almost completely prevented
the mammalian ortholog of the life-extending yeast gene SIR2
in the hyperadiponectinemic mice in association with atten-
are involved in the molecular mechanisms linking lifespan to
uated oxidative DNA damage. The transgenic mice also
adipose tissue. SIRT1 represses peroxisome proliferator-acti-
showed longer life span on a conventional low-fat chow.
vated receptor (PPAR)-c transactivation and inhibits lipid
Adiponectin circulates mainly as a low-molecular-weight
accumulation in adipocytes (113). The favorable effect of ad-
(180 kDa) hexamer and a high-molecular-weight (*360 kDa)
ipose tissue reduction on lifespan could be due to increased
multimer. Adiponectin multimers exert differential biologic
production of anti-inflammatory adipocytokines and de-
effects, with the high-molecular-weight multimer associ-
creased production of pro-inflammatory adipocytokines as
ated with favorable metabolic effects, that is, greater insulin
sensitivity, reduced visceral adipose mass, reduced plasma
clearance of apoptotic cells in advanced plaques (144). Mole-
triglycerides, and increased high-density lipoprotein (HDL)-
cular structure of adiponectin is akin to complement C1q, and
cholesterol (75), and adiponectin knockout mice manifest in-
adiponectin binds to a number of target molecules, including
sulin resistance, IGT, and dyslipidemia (168). Adiponectin,
damaged endothelium and the surface of apoptotic cells (110).
thus, influences atherosclerosis by affecting the balance of
Thus, adiponectin may play a crucial role in efferocytosis and
atherogenic and antiatherogenic lipoproteins in plasma, and
prevention of vulnerable plaque formation. Although a recent
by modulating cellular processes involved in foam cell for-
study showed lack of association between adiponectin levels
mation. The metabolic effects of adiponectin are mediated
and atherosclerosis (99), this attractive hypothesis needs to be
thorough adiponectin receptor-1 and adiponectin receptor-2
explored. The potential roles of adiponectin in prevention of
(68). These adiponectin receptors are linked to AMP-activated
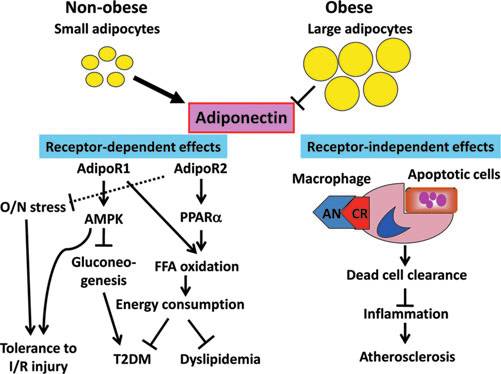
cardiovascular risk are illustrated in Figure 4.
protein kinase (AMPK) and PPAR-a, respectively. AMPK is acellular energy sensor that contributes to the regulation of
energy balance and caloric intake (21, 150). The activity of
Several nonpharmacological interventions can prevent
AMPK is determined by cellular AMP/ATP. AMPK can
endothelial dysfunction or improve impaired endothelium-
phophorylate several enzymes involved in anabolism to
dependent vasodilation. Probably, the most effective non-
prevent further ATP consumption, and induces some cata-
pharmacological measure for the management of MetS is
bolic enzymes to increase ATP generation. Further, AMPK
represented by aerobic physical activity, which can reduce
stimulates glucose utilization in the skeletal muscle and in-
production of oxidative stress associated with increasing age.
hibits gluconeogenesis in the liver (159). On the other hand,
It has been demonstrated that exercise alone is an effective
PPAR-a participates in fatty acid oxidation, thereby increas-
nonpharmacological treatment strategy for insulin resistance,
ing energy consumption.
MetS, and cardiovascular disease risk factors in older obese
Adiponectin is known to enhance ischemic tolerance in the
adults (160). In addition, several randomized, controlled
heart. It has been demonstrated that short-term CR increases
studies have shown that aerobic types of exercise are protec-
adiponectin levels and exerts a cardioprotective effect againstischemia/reperfusion injury in the wild-type mouse but notadiponectin antisense transgenic mouse heart (131), suggest-ing that adiponectin is an obligatory mediator of CR-inducedischemic tolerance in the heart. This cardioprotective effect ofadiponectin is mediated by AMPK-mediated signaling. Pro-longed CR also confers ischemic tolerance, but this effect isindependent of AMPK and mediated by a NO-dependentincrease in nuclear Sirt1 (130), which is responsible for a NAD-dependent deacetylase and prevention of apoptosis in cardiacmyocytes (3). Thus, CR increases ischemic tolerance viaadiponectin- and Sirt1-dependent mechanisms.
Cardioprotection by adiponectin is at least in part mediated
by an antioxidative/nitrosative effect. The recent study hasdemonstrated that adiponectin reduces oxidative/nitrosativestress by inhibiting NADPH oxidase and iNOS expressionand ameliorates ischemia/reperfusion injury in mice (139),and this action is AMPK independent (154). A recent study(86) using a rat model of nonalcoholic steatohepatitis suggeststhat this antioxidative effect may be mediated by adiponectinreceptor-2.
Potential roles of adiponectin in the prevention of
Adiponectin also acts as an anti-inflammatory molecule
cardiovascular risk factors. Small adipocytes in the visceral
through a receptor-independent mechanism. The serum
adipose tissue in nonobese subjects increase generation of adi-ponectin through adiponectin receptor-1 (AdipoR1) and adi-
concentration of adiponectin exceeds a micromolar level that
ponectin receptor-2 (AdipoR2)-dependent and -independent
is extremely higher than estimated its receptor density. Thus,
mechanisms. AdipoR1 improves insulin resistance and inhibits
receptor-independent mechanism has been implicated in
the development of type-2 diabetes mellitus (T2DM) by in-
the anti-inflammatory action of adiponectin. This anti-
hibiting gluconeogenesis in the liver and stimulating glucose
inflammatory effect may play a crucial role in preventing
utilization in the skeletal muscle through the action of AMP-
the development of atherosclerosis and vulnerable plaque.
kinase (AMPK). The AdipoR1-AMPK signaling also confers
Plaque necrosis arises from a combination of foam cell apo-
tolerance to ischemia/reperfusion (I/R) injury. AdipoR2 in-
ptosis and defective clearance of these dead cells, a process
creases free fatty acid (FFA) oxidation and energy consumption
called efferocytosis (137). Defective efferocytosis contributes
though the activation of peroxisome proliferator activated re-
to necrotic core and the vulnerable plaque formation within
ceptor-a (PPAR-a), thereby preventing T2DM and dyslipide-mia. AdipoR2 may also be involved in tolerance to I/R injury
advanced atheroma that is thought to promote plaque dis-
by inhibiting oxidative/nitrosative (O/N) stress (dotted line). A
ruption and, ultimately, acute atherothrombotic vascular
receptor-independent action of adiponectin (AN) is involved in
disease (144). Molecular-genetic causation studies in mouse
an antiatherosclerotic effect through dead cell clearance and
models of advanced atherosclerosis have provided evidence
inhibition of inflammation by acting as a complement for
that several molecules known to be involved in efferocytosis,
macrophages to eliminate apoptotic cells from the atheroma-
including complement C1q, play important roles in the
tous plaque.
OXIDATIVE STRESS AND METABOLIC SYNDROME
adiponectin levels (6, 83, 101). Conversely, exercise trainingmay influence pro-inflammatory cytokine production (18, 46,148). Future studies are needed to investigate the cellularmechanisms by which exercise training affects inflamma-tion and whether alterations in inflammation are one mecha-nism by which exercise improves components of MetS inat-risk individuals.
Antagonists against RAAS
Any anti-HTN therapies have been shown to reduce the
risk of total major cardiovascular events. Recently, the rele-vance of the type of anti-HTN therapy used to treat HTNpatients in facilitating the development of T2DM has beendemonstrated in different trials. The recognition of the riskpresent in HTN patients with MetS for developing T2DMreinforces the need to consider the ideal anti-HTN therapy,either mono or combination, in these patients. The available
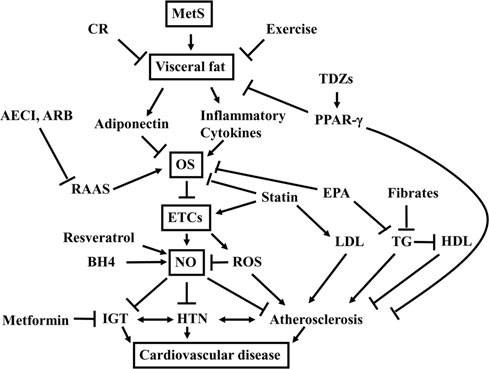
Schematic drawing of therapeutic opportunities
evidence showing that an ACE inhibitor or an ARB is the most
for prevention of cardiovascular disease in MetS. MetS is
suitable therapy to be started in these patients, alone or in
characterized by the cluster of cardiovascular risk factors:
combination, due to their capacity to prevent or retard the
impaired glucose tolerance (IGT), hypertension (HTN), and
development of T2DM (48, 124).
dyslipidemia that culminate in cardiovascular disease. Re-duction of visceral fat by caloric restriction (CR) and exercise
Adipocytes are a suggested source of components of the
increases adiponectin and decreases inflammatory cytokines,
RAAS, with regulation of their production related to obesity-
thereby inhibiting systemic inflammation and oxidative
HTN (22). Ang II has been demonstrated to promote oxidative
stress (OS) to endothelial cells (ETCs). Inhibition of the renin-
stress via overexpression of NADPH oxidase in adypocytes
angiotensin-aldosterone system (RAAS) by employing an
(50, 66). It has been demonstrated that blockade of Ang II
angiotensin converting inhibitor (ACEI) or angiotensin II
type-1 receptors reduces oxidative stress in adipose tissue and
type-1 receptor blocker (ARB) is more direct approach to
ameliorates adipocytokine dysregulation (74). Therefore,
inhibit OS to ETCs. Antidyslipidemic agents, statin and ei-
ACE inhibitors and ARBs represent promising tools for in-
cosapentaenoic acid (EPA), exert ETC protection indepen-
hibiting oxidative stress in adipocytes, thereby preventing the
dent of their effects on LDL and triglyceride (TG) levels.
production of pro-inflammatory adipokines responsible for
Fibrates, on the other hand, decrease TG and increase high-density lipoprotein (HDL), thereby preventing atherosclero-
systemic inflammation and oxidative stress in MetS.
sis. Although OS to ETCs promotes further generation ofROS that inhibit bioavailability of NO by generating perox-
Insulin sensitizers
ynitrite or causing uncoupling of NO synthase, ETC-derived
There are a number of pharmacological tools for treatment
NO synthesis can be increased by resveratrol and an NO
of T2DM. Ever since insulin was discovered in the early 20th
synthase cofactor tetrahydrobiopterin (BH4). Antidiabeticagents such as metformin and thiazolidinediones (TDZs) that
century, it had been an only drug in patients with insulin-
act through an increase in insulin sensitivity improve not
dependent DM and T2DM for many years. Then, sulfonyl
only IGT but also hyperinsulinemia responsible for athero-
urea became available in the mid 20th century, and it had been
sclerosis and cardiovascular disease.
a first choice of drugs in patients with T2DM. However, rec-ognition of deleterious cardiovascular effects of hyper-
tive against age-related increases in visceral adiposity in
insulinemia in patients with T2DM has shifted paradigm of
growing children and adolescents (72). Moreover, physical
T2DM treatment from increasing blood insulin level to insulin
activity can improve endothelial dysfunction even in patients
sensitivity. Insulin acts as not only blood glucose-lowering
with cardiovascular risk factors such as essential HTN. It is
hormone but also acts as a growth factor under oxidative
worth noting that most of nonpharmacological measures for
stress that may be involved in atherosclerosis. Ruige and as-
prevention of cardiovascular risk act by preventing or reducing
sociates (119) have demonstrated that hyperinsulinemia is an
inflammation and oxidative stress. Current evidence supports
independent risk factor of coronary artery disease. In addi-
that aerobic exercise, alone or combined with CR, improves
tion, insulin activates the PI3K-Akt axis that is known to
symptoms of MetS, possibly by altering systemic levels of in-
play a role in the control of aging (9, 109), thereby possibly
flammatory adipocytokines (162). A number of studies show
restricting life span. On the contrary, pharmacological inter-
that increased physical activity leads to lower circulating levels
ventions that increase insulin sensitivity reduce cardiovas-
of pro-inflammatory cytokines and higher levels of adipo-
cular complications and are, therefore, expected to promote
nectin. The mechanism underlying reduced oxidative stress in
longevity. A PPAR-c activator TZD and metformin are quite
visceral white adipose tissue by exercise may be related to the
promising tools to substantially improve the cluster of car-
decreased expression of NADPH oxidase in addition to an
diovascular risk factors in patients with MetS complicated
enhanced antioxidant defense system, and the prevention of
with T2DM, whereas a-glucosidase inhibitors may also
dysregulated production of inflammation-related adipocyto-
be effective to prevent hyperinsulinemia by inhibiting
kines (121), suggesting that exercise is a fundamental approach
postprandial hyperglycemia. A newly emerged antidiabetic
to protect against cardiovascular risk in MetS. However, lim-
drug dipeptidyl peptidase-IV (DPP-4) inhibitors increase
ited data show that exercise training does not influence serum
glucose-dependent stimulation of insulin secretion, and unlike
sulfonyl urea it do not cause hypoglycemia or inhibit ATP-
metformin improves endothelial functions in Otsuka Long-
sensitive potassium channels that are thought to be crucial in
Evans Tokushima fatty rat mesenteric arteries by suppressing
cytoprotection in both pancreatic islet b-cells and cardiomyo-
vasoconstrictor prostanoids and by reducing oxidative stress
cytes (39, 114). Although DPP-4 inhibitors are promising anti-
(85). Metformin confers cardioprotection against ischemia/
diabetic drugs, the cardiovascular effect of the DPP-4 inhibitors
reprfusion injury through a PI3K-mediated inhibition of mi-
in patients with T2DM remains to be investigated, because they
tochondrial permeability transition pore opening (13). In
induce postprandial hyperinsulinemia. Focussed here are two
addition, metformin attenuated oxidative stress-induced
representative insulin sensitizer, TZDs and metformin, because
cardiomyocyte apoptosis and prevented the progression of
these antidiabetic drugs have an established underlying
heart failure in dogs, and this cardioprotective effect was
mechanism for beneficial cardiovascular effects and have
dependent on the activation of AMPK (123). Consistent with
shown strong clinical evidence of reduced cardiovascular risk
the beneficial cardiovascular effect in animals, the UK Pro-
in patients with T2DM.
spective Diabetes Study (144a) showed that metformin de-
The beneficial effect of TDZ is attributed to activation of
creases macrovascular morbidity and mortality independent
PPAR-c. A flurry of human and animal studies has shed a
of glycemic control in patients with T2DM. A subsequent
light on the mechanisms how TZDs act, and which of their
randomized, placebo-controlled trial has demonstrated that
physiological effects are dependent on PPAR-c. It is now ev-
metformin can reduce inflammatory markers and improve
ident that TDZ stimulates PPAR-c in adipocytes in the visceral
endothelial function (35). The potential vascular protective
adipose tissue and increases the generation of adiponectin (24,
effects of metformin may complement other strategies within
126). Further, new roles for PPAR-c signaling beyond the
such a framework. Thus, metformin treatment may represent
metabolic effects through adiponectin have been discovered
a relevant element of an integrated pharmacotherapy to pre-
in inflammation, bone morphogenesis, endothelial function,
vent not only T2DM but also cardiovascular disease in MetS.
cancer, longevity, and atherosclerosis (59). All of the majorcells in the vasculature express PPAR-c, including endothelial
Antidyslipidemic agents
cells, vascular SMCs, and monocytes/macrophages (40, 55).
PPAR-c is present in intimal macrophages in human athero-
Because atherosclerosis is facilitated by dyslipidemia and
mas (5). Recent experimental studies provide evidence that
oxidative stress in patients with T2DM, numerous studies
PPAR-c may function to protect the vasculature from injury
have investigated relative contribution of dyslipidemia and
(81, 103). Cell culture studies have shown that TZD inhibits
oxidative stress to atherogenesis in diabetic animals. It has
both the proliferation and migration of vascular SMCs (105).
been demonstrated that antioxidants vitamin E and probucol
TZDs block vascular SMC growth by inducing cell cycle arrest
and a 3-hydroxy-3-methylglutaryl Co-A reductase inhibitor
in G1 through an inhibition of retinoblastoma protein phos-
lovastatin significantly reduced plasma triglyceride in the
phorylation (80). Migration of monocytes and vascular SMCs
diabetic hamsters fed the atherogenic diet (42). In this study,
is also inhibited by TZDs, possibly through decreased matrix
vitamin E treatment increased total cholesterol, and probucol
metalloproteinase production (108). Activation of PPAR-c by
reduced HDL-cholesterol without affecting total cholesterol,
TZDs in macrophages induces ATP binding cassette trans-
whereas lovastatin reduced total cholesterol and selectively
porter A1 expression to promote reverse cholesterol transport
decreased non-HDL-cholestrerol, and significantly reduced
(102). These effects of PPAR-c culminate in protection of en-
fatty streak lesion formation in the aortic arch. Although vi-
dothelial cells. Thus, TZD activation of PPAR-c may protect
tamin E and probucol were effective in reducing several in-
against atherosclerosis both by normalizing pro-atherogenic
dices of oxidative stress, including plasma lipid peroxides,
metabolic abnormalities of the insulin resistance/diabetes
cholesterol oxidation products, and in vitro LDL oxidation,
milieu and through an inhibition of vascular SMC growth and
they had no effect on fatty streak lesion formation. These re-
movement. Consistent with this hypothesis is the fact that in a
sults indicate that the LDL in diabetic animals is more sus-
large, placebo-controlled, outcome study in secondary pre-
ceptible to oxidation than in nondiabetic hamsters and that
vention, PROactive study, the use of pioglitazone in addition
not only vitamin E and probucol but also lovastatin provide
to an existing optimized macrovascular risk management
antioxidant protection. It appears that in this combined model
resulted in a significant reduction of macrovascular endpoints
of T2DM and hypercholesterolemia, lovastatin prevented
within a short observation period that was comparable to the
progression of fatty streak lesion formation by reducing total
effect of statins and ACE inhibitors in other trials (38). In ad-
cholesterol and non-HDL-cholesterol and inhibiting oxidative
dition, the efficacy of TDZs in preventing atherosclerosis in
patients with T2DM has been confirmed by subsequent clin-
The pleiotropic effects of statins that prevent atherogenesis
ical trials (51). These results underline the value of TDZs for
have been extensively investigated. Emerging evidence sug-
managing the increased cardiovascular risk in MetS compli-
gests that these cholesterol-independent effects are predomi-
cated with T2DM.
nantly due to their ability to inhibit isoprenoid synthesis,
Metformin is widely used as a hypoglycemic reagent for
particularly geranylgeranylpyrophosphate and farnesylpyr-
T2DM. The reduction of hepatic gluconeogenesis is thought to
ophosphate, which are important post-translational lipid at-
be a key effect of metformin, and its molecular mechanism is
tachments of the Rho GTPases and activation of its
attributed to the reduction of glucose-6-phosphatase activity,
downstream target, Rho-kinase (ROCK) (117). Inhibition of
as well as suppression of mRNA expression levels of multiple
ROCK by statins may also be associated with inhibition of
genes linked to the metabolic pathways involved in glucose
oxidative stress mediated by activation of NADPH oxidase. It
and lipid metabolism in the liver (60). However, metformin
has been shown that rosuvastatin attenuated the Ang II-
exerts cardiovascular protection independent of the blood
mediated upregulation of NAPDH oxidase subunits as well as
glucose-lowering efficacy. It has been demonstrated that
nuclear factor-kappaB associated with downregulation of
OXIDATIVE STRESS AND METABOLIC SYNDROME
Ang II type-1 receptors and the lectin-like oxidized LDL re-
that resveratrol confers protection against ischemia/reperfu-
ceptor LOX-1, leading to the reduction of oxidized LDL (70).
sion injury through its antioxidant activity and upregulation of
In this regard, while increased ROCK activity is associated
NO production (56, 63, 116). Moreover, resveratrol modulates
with endothelial dysfunction, cerebral ischemia and coronary
vascular cell function, inhibits LDL oxidation, and suppresses
vasospasms in MetS, the inhibition of ROCK by statins leads
platelet aggregation (17). Miatello and associates (89) demon-
to upregulation of eNOS, decreased vascular inflammation,
strated that chronic administration of resveratrol prevented
and reduced atherosclerotic plaque formation (165).
atherosclerosis in rats, and raised the hypothesis that the in-
In MetS, increased triglyceride in conjunction with elevated
crease in eNOS activity may contribute to the protective
LDL plays a crucial role in atherogenesis. Such combined
properties of resveratrol against cardiovascular disease. Re-
dyslipidemia often requires multiple antidyslipidemic agents.
sults from other laboratories support the unifying hypothesis
Fibrates effectively reduce fasting and postprandial hyper-
that the improvements in risk factors by resveratrol are medi-
triglycemia, shift the distribution of LDL particles toward less
ated by eNOS (111, 118, 164). These results suggest that an
dense particles, and increase HDL-cholesterol. The finding of
adequate supplementation of resveratrol might help to prevent
triglyceride-rich lipoproteins in human atheroma has pro-
or delay the occurrence of atherogenic cardiovascular disease
vided substantial pathophysiologic evidence for a direct role
associated with insulin-resistant states in MetS. In addition,
of triglyceride in atherogenesis (25, 58). Thus, fibrates repre-
recent data provide interesting insights into the effect of re-
sent particularly important tools to manage dyslipidemia in
sveratrol on the lifespan of simple eukaryotes such as yeast and
MetS complicated with T2DM. Indeed, compelling evidence
flies by activating the longevity genes and has been suggested
from meta-analysis of a number of clinical studies on a large
as a CR mimetic (33, 62, 136, 155), implicating the potential of
aggregate of patients has established an increased level of
resveratrol as an antiaging agent in treating age-related human
triglycerides as an independent risk factor for atherosclerotic
diseases. This attractive property of resveratrol against ath-
coronary heart disease (8, 52, 100). However, combination of
erosclerosis and aging should be studied in human especially
statins and fibrates is often contraindicated by increased in-
in patients with MetS. However, the phenolic compound pos-
cidence of myopathy. On the other hand, niacin, fibrates, and
sesses a low bioavailability and rapid clearance from the
bile acid sequestrants are effective in combination with statins
plasma (29). Thus, bioavailability, metabolism, and tissue dis-
in lowering LDL, triglycerides, and total cholesterol levels and
tribution of resveratrol in humans need to be clearly estab-
increasing HDL. Niacin-statin therapy reduces atherosclerotic
lished to develop better biological effects.
progression and coronary events (10, 30, 34, 156). o-3 poly-
Another potential pharmacological tool for the manage-
unsaturated fatty acids, which are abundant in fish oil, are
ment of cardiovascular risk factors in MetS is BH4. BH4 is a
another promising tool for combination therapy for dyslipi-
cofactor of eNOS, iNOS, and neuronal NOS and necessary for
demic patients. The Japan Eicosapentaenoic Acid (EPA) Lipid
NO biosynthesis. Lack of BH4 is associated with uncoupling
Intervention Study demonstrated that EPA prevented major
of NOS, leading to the generation of more superoxide and less
coronary events, including sudden cardiac death, fatal and
NO that shifts the nitroso-redox balance and may have ad-
nonfatal myocardial infarction, and other nonfatal events,
verse consequences on cardiovascular function. This trans-
including unstable angina pectoris, angioplasty, stenting, or
formation of NOS especially eNOS from a protective enzyme
coronary artery bypass grafting in hypercholesterolemic pa-
to a contributor to oxidative stress has been observed in sev-
tients (161). The beneficial effect of EPA significantly corre-
eral in vitro models, in animal models of cardiovascular dis-
lated with the reduction of triglyceride and the increase in
ease, and in patients with cardiovascular risk factors (45, 90,
HDL (120), although cardiovascular protection by EPA may
91). BH4 is highly sensitive to oxidation by ROS and perox-
also be attributed to the anti-inflammatory effect and inhibi-
ynitrite and is converted to dihydrobiopterin (BH2). Oxida-
tion of platelet function (93, 112). Taken all together, both
tive stress imposed on endothelial cells causes depletion of
triglyceride and HDL levels correlate with cardiovascular risk
BH4 and eNOS uncoupling. In many cases, supplementation
and should be considered secondary targets of therapy.
with BH4 under pathological conditions with oxidative stress
Combination therapy can be safe and effective and can be
has been shown to correct eNOS dysfunction in animal
constructed to affect all lipoprotein parameters. However,
models and patients (45, 90, 91). However, true mechanistic
studies still are needed showing definite evidence on differ-
relationship between endothelial BH4 levels and eNOS reg-
ential therapy in lipid lowering based on prospective, con-
ulation in vivo by administration of BH4 remains controver-
trolled trials with endpoints of macro- and microangiopathy
sial. High extracellular BH4 concentrations may result in
in MetS complicated with T2DM and dyslipidemia.
nonspecific antioxidant effects that indirectly increase NObioactivity by ROS scavenging rather than by modulation ofeNOS activity. Further, the effects of supplementation with
Other potential pharmacological tools
BH4 or biopterin analogs on NO bioactivity are unpredictable
Epidemiological studies suggest that the consumption of
in vascular disease states in which oxidative stress is increased
wine, particularly of red wine, reduces the incidence of mor-
(140, 146). Indeed, it remains unclear whether adequate eNOS
tality and morbidity from coronary heart disease. This has
cofactor function in vivo is related to absolute BH4 levels in the
given rise to what is now popularly termed the ‘‘French
endothelial cell, or whether the relative balance between re-
paradox'' (28). The cardioprotective effect has been attributed
duced BH4 and oxidized BH2 may be more important (147).
to antioxidants present in the polyphenol fraction of red wine.
Intracellular BH4 levels are regulated by the activity of the
Grapes contain a variety of antioxidants, including resveratrol,
de novo biosynthetic pathway and the salvage pathway. In the
catechin, epicatechin, and proanthocyanidins. Of these, re-
de novo biosynthetic pathway, guanosine triphosphate cyclo-
sveratrol is present mainly in grape skin, whereas proantho-
hydrolase (GTPCH)-1 catalyzes GTP to dihydroneopterin
cyanidin is present in the seeds. Emerging evidence indicates
triphosphate. BH4 is generated by further steps catalyzed by
6-pyruvoyltetrahydropterin synthase and sepiapterin reduc-
ture of Japan and Promotion and Mutual Aid Corporation for
tase (142). GTPCH-1 appears to be the rate-limiting enzyme in
Private Schools of Japan.
BH4 biosynthesis; transgenic overexpression of GTPCH-1 issufficient to augment BH4 levels in endothelial cells and
preserve NO-mediated endothelial function in diabetic mice(4). In the salvage pathway, BH4 is synthesized from BH2 by
1. This reference has been deleted.
sepiapterin reductase and dihydrofolate reductase. Exogen-
2. Alba AC and Delgado DH. Optimal medical treatment of
cardiovascular risk factors: can we prevent the develop-
ous BH4 is labile in physiological solution. It has been re-
ment of heart failure? Expert Rev Cardiovasc Ther 7: 147–
ported that in vivo half-life of BH4 is 3.3–5.1 h in the plasma of
healthy adult humans (44). Because not all oxidized BH4 is
3. Alcendor RR, Kirshenbaum LA, Imai S, Vatner SF, and
converted to BH2, which is further degraded to dihydrox-
Sadoshima J. Silent information regulator 2alpha, a lon-
anthopterin and excreted to urine (129), BH2 availability for
gevity factor and class III histone deacetylase, is an essen-
the salvage pathway may be limited under oxidative stress
tial endogenous apoptosis inhibitor in cardiac myocytes.
even with BH4 supplementation. Thus, sepiapterin may serve
Circ Res 95: 971–980, 2004.
as an effective substrate for BH4 via the salvage pathway. Folic
4. Alp NJ, Mussa S, Khoo J, Cai S, Guzik T, Jefferson A, Goh
acid and vitamin C are also able to restore eNOS functionality,
N, Rockett KA, and Channon KM. Tetrahydrobiopterin-
most probably by enhancing BH4 levels through mechanisms
dependent preservation of nitric oxide-mediated endo-
yet to be clarified (129). The therapeutic efficacy of BH4 has
thelial function in diabetes by targeted transgenic
been examined in patients with HTN, peripheral arterial dis-
GTP-cyclohydrolase I overexpression. J Clin Invest 112:
ease, and coronary artery disease, and these studies consis-
725–735, 2003.
tently demonstrated the beneficial effect of BH4 on endothelial
5. Amoruso A, Bardelli C, Fresu LG, Palma A, Vidali M,
dysfunction (71). However, a phase-2 clinical trial sponsored
Ferrero V, Ribichini F, Vassanelli C, and Brunelleschi S.
by the U.S. pharmaceutical company BioMarin failed to ob-
serve an ameliorative effect of oral administration of BH4 in
gamma expression in monocyte/macrophages from coro-
patients with poorly controlled HTN. Further studies are
nary artery disease patients and possible gender differences.
needed to address whether BH4 or its analogs truly exert sal-
J Pharmacol Exp Ther 331: 531–538, 2009.
utary effects on endothelial dysfunction induced by a variety
6. Ando D, Hosaka Y, Suzuki K, and Yamagata Z. Effects of
of vascular disease. Therapeutic opportunities for prevention
exercise training on circulating high molecular weightadiponectin and adiponectin oligomer composition: a ran-
of cardiovascular disease in MetS are illustrated in Figure 5.
domized controlled trial. J Atheroscler Thromb 16: 733–739,2009.
Concluding Remarks
7. Archuleta TL, Lemieux AM, Saengsirisuwan V, Teachey
Abdominal obesity is a cause of all the morbidity of MetS.
MK, Lindborg KA, Kim JS, and Henriksen EJ. Oxidant
Oxidative stress develops in hypertrophied adipocytes, which
stress-induced loss of IRS-1 and IRS-2 proteins in rat skel-
increase the synthesis of pro-inflammatory cytokines, while
etal muscle: role of p38 MAPK. Free Radic Biol Med 47:
decreasing anti-inflammatory cytokines. Dysregulation of
1486–1493, 2009.
such adipocytokines is responsible for systemic inflammation
8. Avogaro A, Giorda C, Maggini M, Mannucci E, Raschetti R,
and oxidative stress and contributes to the pathogenesis of the
Lombardo F, Spila-Alegiani S, Turco S, Velussi M, and
obesity-associated morbidity in MetS. Decrease in abdominal
Ferrannini E. Incidence of coronary heart disease in type 2diabetic men and women: impact of microvascular com-
obesity by lifestyle interventions is fundamental approach to
plications, treatment, and geographic location. Diabetes Care
MetS. However, CR and exercise are often difficult in patients
30: 1241–1247, 2007.
with MetS. Thus, alternative strategies are required to prevent
9. Bartke A. Impact of reduced insulin-like growth factor-1/
cardiovascular risk in MetS. Accumulating basic research
insulin signaling on aging in mammals: novel findings.
evidence indicates that endothelial cells are primarily affected
Aging Cell 7: 285–290, 2008.
by inflammation and become a source of further oxidative
10. Bays H. Safety of niacin and simvastatin combination
stress in the vascular wall and surrounding cells, leading to
therapy. Am J Cardiol 101: 3B–8B, 2008.
IGT, HTN, and atherosclerosis. Thus, the endothelium is
11. Bendall JK, Alp NJ, Warrick N, Cai S, Adlam D, Rockett K,
recognized as a major therapeutic target in the prevention and
Yokoyama M, Kawashima S, and Channon KM. Stoichio-
treatment of vascular disease in patients with MetS. The
purpose of improving endothelial function is to restore nor-
biopterin, endothelial NO synthase (eNOS) activity, and
mal biosynthesis of NO and the reduction of excessive gen-
eNOS coupling in vivo: insights from transgenic mice with
eration of ROS. Currently available pharmacological tools
endothelial-targeted GTP cyclohydrolase 1 and eNOS over-
such as ACE inhibitors, ARBs, TDZs, metformin, and statins
expression. Circ Res 97: 864–871, 2005.
are effective in preventing cardiovascular risk in MetS
12. Berg AH and Scherer PE. Adipose tissue, inflammation,
through reduction of inflammation and oxidative stress either
and cardiovascular disease. Circ Res 96: 939–949, 2005.
in the visceral adipose tissue or endothelial cells. Further
13. Bhamra GS, Hausenloy DJ, Davidson SM, Carr RD, Paiva
studies are needed to develop more effective strategy to
M, Wynne AM, Mocanu MM, and Yellon DM. Metformin
manage cardiovascular risk in MetS.
protects the ischemic heart by the Akt-mediated inhibitionof mitochondrial permeability transition pore opening.
Basic Res Cardiol 103: 274–284, 2008.
14. Bitar MS, Al-Saleh E, and Al-Mulla F. Oxidative stress—
This work was supported in part by Research Grant
mediated alterations in glucose dynamics in a genetic ani-
20590847 from the Ministry of Education, Science, and Cul-
mal model of type II diabetes. Life Sci 77: 2552–2573, 2005.
OXIDATIVE STRESS AND METABOLIC SYNDROME
15. Bluher M. Fat tissue and long life. Obes Facts 1: 176–182,
34. Davidson MH and Toth PP. Combination therapy in the
management of complex dyslipidemias. Curr Opin Lipidol
16. Boyle JJ. Macrophage activation in atherosclerosis: patho-
15: 423–431, 2004.
genesis and pharmacology of plaque rupture. Curr Vasc
35. De Jager J, Kooy A, Lehert P, Bets D, Wulffele MG, Teerlink
Pharmacol 3: 63–68, 2005.
T, Scheffer PG, Schalkwijk CG, Donker AJ, and Stehouwer
17. Bradamante S, Barenghi L, and Villa A. Cardiovascular
CD. Effects of short-term treatment with metformin on
protective effects of resveratrol. Cardiovasc Drug Rev 22:
markers of endothelial function and inflammatory activity
169–188, 2004.
in type 2 diabetes mellitus: a randomized, placebo-con-
18. Bradley RL, Jeon JY, Liu FF, and Maratos-Flier E. Voluntary
trolled trial. J Intern Med 257: 100–109, 2005.
exercise improves insulin sensitivity and adipose tissue
36. Dickhout JG, Hossain GS, Pozza LM, Zhou J, Lhotak S, and
inflammation in diet-induced obese mice. Am J Physiol
Austin RC. Peroxynitrite causes endoplasmic reticulum
Endocrinol Metab 295: E586–E594, 2008.
stress and apoptosis in human vascular endothelium: im-
19. Brozinick JT, Jr., Roberts BR, and Dohm GL. Defective
plications in atherogenesis. Arterioscler Thromb Vasc Biol 25:
signaling through Akt-2 and - 3 but not Akt-1 in insulin-
2623–2629, 2005.
resistant human skeletal muscle: potential role in insulin
37. DiCorleto PE. Cellular mechanisms of atherogenesis. Am J
resistance. Diabetes 52: 935–941, 2003.
Hypertens 6: 314S–318S, 1993.
20. Bulcao C, Ferreira SR, Giuffrida FM, and Ribeiro-Filho FF.
38. Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E,
The new adipose tissue and adipocytokines. Curr Diabetes
Massi-Benedetti M, Moules IK, Skene AM, Tan MH, Le-
Rev 2: 19–28, 2006.
febvre PJ, Murray GD, Standl E, Wilcox RG, Wilhelmsen L,
21. Carling D, Sanders MJ, and Woods A. The regulation of
Betteridge J, Birkeland K, Golay A, Heine RJ, Koranyi L,
AMP-activated protein kinase by upstream kinases. Int J
Laakso M, Mokan M, Norkus A, Pirags V, Podar T, Scheen
Obes (Lond) 32 Suppl 4: S55–S59, 2008.
A, Scherbaum W, Schernthaner G, Schmitz O, Skrha J,
22. Cassis LA, Police SB, Yiannikouris F, and Thatcher SE.
Smith U, and Taton J. Secondary prevention of macro-
Local adipose tissue renin-angiotensin system. Curr Hy-
vascular events in patients with type 2 diabetes in the
pertens Rep 10: 93–98, 2008.
PROactive Study (PROspective pioglitAzone Clinical Trial
23. Chen B, Wei J, Wang W, Cui G, Zhao Y, Zhu X, Zhu M,
In macroVascular Events): a randomised controlled trial.
Guo W, and Yu J. Identification of signaling pathways in-
Lancet 366: 1279–1289, 2005.
volved in aberrant production of adipokines in adipocytes
39. Doupis J and Veves A. DPP4 inhibitors: a new approach in
undergoing oxidative stress. Arch Med Res 40: 241–248,
diabetes treatment. Adv Ther 25: 627–643, 2008.
40. Duan SZ, Usher MG, and Mortensen RM. Peroxisome
24. Choi KC, Ryu OH, Lee KW, Kim HY, Seo JA, Kim SG, Kim
proliferator-activated receptor-gamma-mediated effects in
NH, Choi DS, Baik SH, and Choi KM. Effect of PPAR-alpha
the vasculature. Circ Res 102: 283–294, 2008.
and -gamma agonist on the expression of visfatin, adipo-
41. Duda DG, Fukumura D, and Jain RK. Role of eNOS in
nectin, and TNF-alpha in visceral fat of OLETF rats. Bio-
neovascularization: NO for endothelial progenitor cells.
chem Biophys Res Commun 336: 747–753, 2005.
Trends Mol Med 10: 143–145, 2004.
25. Choy PC, Siow YL, Mymin D, and O K. Lipids and ath-
42. El-Swefy S, Schaefer EJ, Seman LJ, van Dongen D, Sevanian
erosclerosis. Biochem Cell Biol 82: 212–224, 2004.
A, Smith DE, Ordovas JM, El-Sweidy M, and Meydani M.
26. Collot-Teixeira S, Martin J, McDermott-Roe C, Poston R,
The effect of vitamin E, probucol, and lovastatin on oxi-
and McGregor JL. CD36 and macrophages in atheroscle-
dative status and aortic fatty lesions in hyperlipidemic-
rosis. Cardiovasc Res 75: 468–477, 2007.
diabetic hamsters. Atherosclerosis 149: 277–286, 2000.
27. Colman RJ, Anderson RM, Johnson SC, Kastman EK,
43. Evans JL, Maddux BA, and Goldfine ID. The molecular
Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons
basis for oxidative stress-induced insulin resistance. Anti-
HA, Kemnitz JW, and Weindruch R. Caloric restriction
oxid Redox Signal 7: 1040–1052, 2005.
delays disease onset and mortality in rhesus monkeys.
44. Fiege B, Ballhausen D, Kierat L, Leimbacher W, Goriounov
Science 325: 201–204, 2009.
D, Schircks B, Thony B, and Blau N. Plasma tetra-
28. Constant J. Alcohol, ischemic heart disease, and the French
hydrobiopterin and its pharmacokinetic following oral
paradox. Coron Artery Dis 8: 645–649, 1997.
administration. Mol Genet Metab 81: 45–51, 2004.
29. Cottart CH, Nivet-Antoine V, Laguillier-Morizot C, and
45. Forstermann U and Munzel T. Endothelial nitric oxide
Beaudeux JL. Resveratrol bioavailability and toxicity in
synthase in vascular disease: from marvel to menace. Cir-
humans. Mol Nutr Food Res 54: 7–16.
culation 113: 1708–1714, 2006.
30. Cziraky MJ, Watson KE, and Talbert RL. Targeting low
46. Forsythe LK, Wallace JM, and Livingstone MB. Obesity and
HDL-cholesterol to decrease residual cardiovascular risk in
inflammation: the effects of weight loss. Nutr Res Rev 21:
the managed care setting. J Manag Care Pharm 14: S3–S28;
117–133, 2008.
quiz S30–S21, 2008.
47. Fujioka S, Matsuzawa Y, Tokunaga K, and Tarui S. Con-
31. Dandona P, Dhindsa S, Ghanim H, and Chaudhuri A.
tribution of intra-abdominal fat accumulation to the im-
Angiotensin II and inflammation: the effect of angiotensin-
pairment of glucose and lipid metabolism in human
converting enzyme inhibition and angiotensin II receptor
obesity. Metabolism 36: 54–59, 1987.
blockade. J Hum Hypertens 21: 20–27, 2007.
48. Fukui T, Rahman M, Hayashi K, Takeda K, Higaki J, Sato
32. Das M, Gabriely I, and Barzilai N. Caloric restriction, body
T, Fukushima M, Sakamoto J, Morita S, Ogihara T,
fat and ageing in experimental models. Obes Rev 5: 13–19,
Fukiyama K, Fujishima M, and Saruta T. Candesartan an-
tihypertensive survival evaluation in Japan (CASE-J) trial
33. Dasgupta B and Milbrandt J. Resveratrol stimulates AMP
of cardiovascular events in high-risk hypertensive patients:
kinase activity in neurons. Proc Natl Acad Sci U S A 104:
rationale, design, and methods. Hypertens Res 26: 979–990,
7217–7222, 2007.
49. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada
63. Hung LM, Chen JK, Huang SS, Lee RS, and Su MJ.
Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M,
Cardioprotective effect of resveratrol, a natural antioxi-
and Shimomura I. Increased oxidative stress in obesity and
dant derived from grapes. Cardiovasc Res 47: 549–555,
its impact on metabolic syndrome. J Clin Invest 114: 1752–
64. Hung YC, Hong MY, and Huang GS. Cholesterol loading
50. Gao YJ, Takemori K, Su LY, An WS, Lu C, Sharma AM, and
augments oxidative stress in macrophages. FEBS Lett 580:
Lee RM. Perivascular adipose tissue promotes vasocon-
849–861, 2006.
striction: the role of superoxide anion. Cardiovasc Res 71:
65. Igarashi M, Hirata A, Yamaguchi H, Tsuchiya H, Ohnuma
363–373, 2006.
H, Tominaga M, Daimon M, and Kato T. Candesartan in-
51. Goldberg RB. The new clinical trials with thiazolidine-
hibits carotid intimal thickening and ameliorates insulin
diones—DREAM, ADOPT, and CHICAGO: promises ful-
resistance in balloon-injured diabetic rats. Hypertension 38:
filled? Curr Opin Lipidol 18: 435–442, 2007.
1255–1259, 2001.
52. Goldenberg I, Benderly M, Sidi R, Boyko V, Tenenbaum A,
66. Inoguchi T and Nawata H. NAD(P)H oxidase activation: a
Tanne D, and Behar S. Relation of clinical benefit of raising
potential target mechanism for diabetic vascular compli-
high-density lipoprotein cholesterol to serum levels of low-
cations, progressive beta-cell dysfunction and metabolic
density lipoprotein cholesterol in patients with coronary
syndrome. Curr Drug Targets 6: 495–501, 2005.
heart disease (from the bezafibrate infarction prevention
67. Jelic S and Le Jemtel TH. Inflammation, oxidative stress,
trial). Am J Cardiol 103: 41–45, 2009.
and the vascular endothelium in obstructive sleep apnea.
53. Goldstein LB, Adams R, Alberts MJ, Appel LJ, Brass LM,
Trends Cardiovasc Med 18: 253–260, 2008.
Bushnell CD, Culebras A, DeGraba TJ, Gorelick PB, Guyton
68. Kadowaki T and Yamauchi T. Adiponectin and adipo-
JR, Hart RG, Howard G, Kelly-Hayes M, Nixon JV, and
nectin receptors. Endocr Rev 26: 439–451, 2005.
Sacco RL. Primary prevention of ischemic stroke: a guide-
69. Kalinowski L and Malinski T. Endothelial NADH/
line from the American Heart Association/American
NADPH-dependent enzymatic sources of superoxide pro-
Stroke Association Stroke Council: cosponsored by the
duction: relationship to endothelial dysfunction. Acta Bio-
chim Pol 51: 459–469, 2004.
disciplinary Working Group; Cardiovascular Nursing
70. Kang BY and Mehta JL. Rosuvastatin attenuates Ang II—
Council; Clinical Cardiology Council; Nutrition, Physical
mediated cardiomyocyte hypertrophy via inhibition of
Activity, and Metabolism Council; and the Quality of Care
LOX-1. J Cardiovasc Pharmacol Ther 14: 283–291, 2009.
and Outcomes Research Interdisciplinary Working Group.
71. Katusic ZS, d'Uscio LV, and Nath KA. Vascular protection
Circulation 113: e873–e923, 2006.
by tetrahydrobiopterin: progress and therapeutic pros-
54. Greenstein AS, Khavandi K, Withers SB, Sonoyama K,
pects. Trends Pharmacol Sci 30: 48–54, 2009.
Clancy O, Jeziorska M, Laing I, Yates AP, Pemberton PW,
72. Kim Y and Lee S. Physical activity and abdominal obesity
Malik RA, and Heagerty AM. Local inflammation and
in youth. Appl Physiol Nutr Metab 34: 571–581, 2009.
hypoxia abolish the protective anticontractile properties of
73. Koh KK, Oh PC, and Quon MJ. Does reversal of oxidative
perivascular fat in obese patients. Circulation 119: 1661–
stress and inflammation provide vascular protection? Car-
diovasc Res 81: 649–659, 2009.
55. Hamblin M, Chang L, Fan Y, Zhang J, and Chen YE. PPARs
74. Kurata A, Nishizawa H, Kihara S, Maeda N, Sonoda M,
and the cardiovascular system. Antioxid Redox Signal 11:
Okada T, Ohashi K, Hibuse T, Fujita K, Yasui A, Hiuge A,
1415–1452, 2009.
Kumada M, Kuriyama H, Shimomura I, and Funahashi T.
56. Hattori R, Otani H, Maulik N, and Das DK. Pharmaco-
Blockade of angiotensin II type-1 receptor reduces oxida-
logical preconditioning with resveratrol: role of nitric
tive stress in adipose tissue and ameliorates adipocytokine
oxide. Am J Physiol Heart Circ Physiol 282: H1988–H1995,
dysregulation. Kidney Int 70: 1717–1724, 2006.
75. Lara-Castro C, Luo N, Wallace P, Klein RL, and Garvey
57. Hattori Y, Akimoto K, Gross SS, Hattori S, and Kasai K.
WT. Adiponectin multimeric complexes and the metabolic
Angiotensin-II-induced oxidative stress elicits hypoadipo-
syndrome trait cluster. Diabetes 55: 249–259, 2006.
nectinaemia in rats. Diabetologia 48: 1066–1074, 2005.
76. Lau DC, Dhillon B, Yan H, Szmitko PE, and Verma S.
58. Heeren J, Beisiegel U, and Grewal T. Apolipoprotein E re-
Adipokines: molecular links between obesity and ather-
cycling: implications for dyslipidemia and atherosclerosis.
oslcerosis. Am J Physiol Heart Circ Physiol 288: H2031–
Arterioscler Thromb Vasc Biol 26: 442–448, 2006.
H2041, 2005.
59. Heikkinen S, Auwerx J, and Argmann CA. PPARgamma in
77. Lavie L, and Lavie P. Molecular mechanisms of cardio-
human and mouse physiology. Biochim Biophys Acta 1771:
vascular disease in OSAHS: the oxidative stress link. Eur
999–1013, 2007.
Respir J 33: 1467–1484, 2009.
60. Heishi M, Ichihara J, Teramoto R, Itakura Y, Hayashi K,
78. Le Brocq M, Leslie SJ, Milliken P, and Megson IL. En-
Ishikawa H, Gomi H, Sakai J, Kanaoka M, Taiji M, and
dothelial dysfunction: from molecular mechanisms to
Kimura T. Global gene expression analysis in liver of obese
measurement, clinical implications, and therapeutic op-
diabetic db/db mice treated with metformin. Diabetologia
portunities. Antioxid Redox Signal 10: 1631–1674, 2008.
49: 1647–1655, 2006.
79. Libby P. The molecular mechanisms of the thrombotic
61. Holloszy JO and Fontana L. Caloric restriction in humans.
complications of atherosclerosis. J Intern Med 263: 517–527,
Exp Gerontol 42: 709–712, 2007.
62. Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu
80. Lim HJ, Lee S, Park JH, Lee KS, Choi HE, Chung KS,
S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL,
Lee HH, and Park HY. PPAR delta agonist L-165041 in-
Scherer B, and Sinclair DA. Small molecule activators of
hibits rat vascular smooth muscle cell proliferation and
sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425:
migration via inhibition of cell cycle. Atherosclerosis 202:
191–196, 2003.
446–454, 2009.
OXIDATIVE STRESS AND METABOLIC SYNDROME
81. Lim S, Jin CJ, Kim M, Chung SS, Park HS, Lee IK, Lee CT,
98. Napoli C, de Nigris F, Williams-Ignarro S, Pignalosa O,
Cho YM, Lee HK, and Park KS. PPARgamma gene transfer
Sica V, and Ignarro LJ. Nitric oxide and atherosclerosis: an
sustains apoptosis, inhibits vascular smooth muscle cell
update. Nitric Oxide 15: 265–279, 2006.
proliferation, and reduces neointima formation after bal-
99. Nawrocki AR, Hofmann SM, Teupser D, Basford JE, Dur-
loon injury in rats. Arterioscler Thromb Vasc Biol 26: 808–813,
and JL, Jelicks LA, Woo CW, Kuriakose G, Factor SM,
Tanowitz HB, Hui DY, Tabas I, and Scherer PE. Lack of
82. Luque Contreras D, Vargas Robles H, Romo E, Rios A, and
association between adiponectin levels and atherosclerosis
Escalante B. The role of nitric oxide in the post-ischemic
in mice. Arterioscler Thromb Vasc Biol 30: 1159–1165, 2010.
revascularization process. Pharmacol Ther 112: 553–563,
100. Nesto RW. Beyond low-density lipoprotein: addressing the
atherogenic lipid triad in type 2 diabetes mellitus and the
83. Magkos F, Mohammed BS, and Mittendorfer B. Enhanced
metabolic syndrome. Am J Cardiovasc Drugs 5: 379–387,
insulin sensitivity after acute exercise is not associated with
changes in high-molecular weight adiponectin concentra-
101. Numao S, Suzuki M, Matsuo T, Nomata Y, Nakata Y,
tion in plasma. Eur J Endocrinol 162: 61–66, 2010.
and Tanaka K. Effects of acute aerobic exercise on high-
84. Martinet W and Kockx MM. Apoptosis in atherosclerosis:
molecular-weight adiponectin. Med Sci Sports Exerc 40:
focus on oxidized lipids and inflammation. Curr Opin Li-
1271–1276, 2008.
pidol 12: 535–541, 2001.
102. Ogata M, Tsujita M, Hossain MA, Akita N, Gonzalez FJ,
85. Matsumoto T, Noguchi E, Ishida K, Kobayashi T, Yamada
Staels B, Suzuki S, Fukutomi T, Kimura G, and Yokoyama
N, and Kamata K. Metformin normalizes endothelial
S. On the mechanism for PPAR agonists to enhance ABCA1
function by suppressing vasoconstrictor prostanoids in
gene expression. Atherosclerosis 205: 413–419, 2009.
mesenteric arteries from OLETF rats, a model of type 2
103. Ogawa D, Nomiyama T, Nakamachi T, Heywood EB,
diabetes. Am J Physiol Heart Circ Physiol 295: H1165–H1176,
Stone JF, Berger JP, Law RE, and Bruemmer D. Activation
of peroxisome proliferator-activated receptor gamma sup-
86. Matsunami T, Sato Y, Ariga S, Sato T, Kashimura H, Ha-
presses telomerase activity in vascular smooth muscle cells.
segawa Y, and Yukawa M. Regulation of oxidative stress
Circ Res 98: e50–e59, 2006.
and inflammation by hepatic adiponectin receptor 2 in an
104. Ogihara T, Asano T, Katagiri H, Sakoda H, Anai M, Sho-
animal model of nonalcoholic steatohepatitis. Int J Clin Exp
jima N, Ono H, Fujishiro M, Kushiyama A, Fukushima Y,
Pathol 3: 472–481, 2010.
Kikuchi M, Noguchi N, Aburatani H, Gotoh Y, Komuro I,
87. Maury E and Brichard SM. Adipokine dysregulation, adi-
and Fujita T. Oxidative stress induces insulin resistance by
pose tissue inflammation and metabolic syndrome. Mol Cell
activating the nuclear factor-kappa B pathway and dis-
Endocrinol 314: 1–16, 2010.
rupting normal subcellular distribution of phosphatidyli-
88. Miard S and Picard F. Obesity and aging have diver-
nositol 3-kinase. Diabetologia 47: 794–805, 2004.
gent genomic fingerprints. Int J Obes (Lond) 32: 1873–1874,
105. Okura T, Nakamura M, Takata Y, Watanabe S, Kitami Y,
and Hiwada K. Troglitazone induces apoptosis via the p53
89. Miatello R, Vazquez M, Renna N, Cruzado M, Zumino AP,
and Gadd45 pathway in vascular smooth muscle cells. Eur
and Risler N. Chronic administration of resveratrol pre-
J Pharmacol 407: 227–235, 2000.
vents biochemical cardiovascular changes in fructose-fed
106. Otabe S, Yuan X, Fukutani T, Wada N, Hashinaga T, Na-
rats. Am J Hypertens 18: 864–870, 2005.
kayama H, Hirota N, Kojima M, and Yamada K. Over-
90. Moens AL and Kass DA. Tetrahydrobiopterin and cardio-
expression of human adiponectin in transgenic mice results
vascular disease. Arterioscler Thromb Vasc Biol 26: 2439–
in suppression of fat accumulation and prevention of pre-
mature death by high-calorie diet. Am J Physiol Endocrinol
91. Moens AL and Kass DA. Therapeutic potential of tetra-
Metab 293: E210–E218, 2007.
hydrobiopterin for treating vascular and cardiac disease. J
107. Otani H. Ischemic preconditioning: from molecular mech-
Cardiovasc Pharmacol 50: 238–246, 2007.
anisms to therapeutic opportunities. Antioxid Redox Signal
92. Monroe VS, Kerensky RA, Rivera E, Smith KM, and Pepine
10: 207–247, 2008.
CJ. Pharmacologic plaque passivation for the reduction of
108. Pakala R, Dilcher C, Baffour R, Hellinga D, Seabron R,
recurrent cardiac events in acute coronary syndromes. J Am
Joner M, Kolodgie F, Virmani R, and Waksman R. Peroxi-
Coll Cardiol 41: 23S–30S, 2003.
some proliferator-activated receptor gamma ligand piogli-
93. Mori TA and Beilin LJ. Omega-3 fatty acids and inflam-
tazone alters neointimal composition in a balloon-denuded
mation. Curr Atheroscler Rep 6: 461–467, 2004.
and radiated hypercholesterolemic rabbit. J Cardiovasc
94. Muller G and Morawietz H. Nitric oxide, NAD(P)H oxi-
Pharmacol 48: 299–305, 2006.
dase, and atherosclerosis. Antioxid Redox Signal 11: 1711–
109. Paradis S, Ailion M, Toker A, Thomas JH, and Ruvkun G.
A PDK1 homolog is necessary and sufficient to transduce
95. Muoio DM and Newgard CB. Metabolism: A is for adi-
AGE-1 PI3 kinase signals that regulate diapause in Cae-
pokine. Nature 436: 337–338, 2005.
norhabditis elegans. Genes Dev 13: 1438–1452, 1999.
96. Muzumdar R, Allison DB, Huffman DM, Ma X, Atzmon G,
110. Peake PW, Shen Y, Walther A, and Charlesworth JA.
Einstein FH, Fishman S, Poduval AD, McVei T, Keith SW,
Adiponectin binds C1q and activates the classical pathway
and Barzilai N. Visceral adipose tissue modulates mam-
of complement. Biochem Biophys Res Commun 367: 560–565,
malian longevity. Aging Cell 7: 438–440, 2008.
97. Myllarniemi M, Calderon L, Lemstrom K, Buchdunger E,
111. Penumathsa SV, Thirunavukkarasu M, Koneru S, Juhasz B,
and Hayry P. Inhibition of platelet-derived growth factor
Zhan L, Pant R, Menon VP, Otani H, and Maulik N. Statin
receptor tyrosine kinase inhibits vascular smooth muscle
and resveratrol in combination induces cardioprotection
cell migration and proliferation. FASEB J 11: 1119–1126,
against myocardial infarction in hypercholesterolemic rat. J
Mol Cell Cardiol 42: 508–516, 2007.
112. Phang M, Garg ML, and Sinclair AJ. Inhibition of
127. Shepherd PR and Kahn BB. Glucose transporters and in-
platelet aggregation by omega-3 polyunsaturated fatty
sulin action—implications for insulin resistance and dia-
acids is gender specific-Redefining platelet response to
betes mellitus. N Engl J Med 341: 248–257, 1999.
fish oils. Prostaglandins Leukot Essent Fatty Acids 81: 35–
128. Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Lau-
bach VE, Sherman PA, Sessa WC, and Smithies O. Elevated
113. Picard F and Guarente L. Molecular links between aging
blood pressures in mice lacking endothelial nitric oxide
and adipose tissue. Int J Obes (Lond) 29 Suppl 1: S36–S39,
synthase. Proc Natl Acad Sci U S A 93: 13176–13181, 1996.
129. Shi W, Meininger CJ, Haynes TE, Hatakeyama K, and Wu
114. Quast U, Stephan D, Bieger S, and Russ U. The impact of
G. Regulation of tetrahydrobiopterin synthesis and bio-
ATP-sensitive K + channel subtype selectivity of insulin
availability in endothelial cells. Cell Biochem Biophys 41:
secretagogues for the coronary vasculature and the myo-
415–434, 2004.
cardium. Diabetes 53 Suppl 3: S156–S164, 2004.
130. Shinmura K, Tamaki K, and Bolli R. Impact of 6-mo caloric
115. Rahaman SO, Lennon DJ, Febbraio M, Podrez EA, Hazen
restriction on myocardial ischemic tolerance: possible in-
SL, and Silverstein RL. A CD36-dependent signaling cas-
volvement of nitric oxide-dependent increase in nuclear
cade is necessary for macrophage foam cell formation. Cell
Sirt1. Am J Physiol Heart Circ Physiol 295: H2348–H2355, 2008.
Metab 4: 211–221, 2006.
131. Shinmura K, Tamaki K, Saito K, Nakano Y, Tobe T, and Bolli
116. Ray PS, Maulik G, Cordis GA, Bertelli AA, Bertelli A, and
R. Cardioprotective effects of short-term caloric restriction are
Das DK. The red wine antioxidant resveratrol protects
mediated by adiponectin via activation of AMP-activated
isolated rat hearts from ischemia reperfusion injury. Free
protein kinase. Circulation 116: 2809–2817, 2007.
Radic Biol Med 27: 160–169, 1999.
132. Skultetyova D, Filipova S, Riecansky I, and Skultety J. The
117. Rikitake Y and Liao JK. Rho GTPases, statins, and nitric
role of angiotensin type 1 receptor in inflammation and
oxide. Circ Res 97: 1232–1235, 2005.
endothelial dysfunction. Recent Pat Cardiovasc Drug Discov
118. Rivera L, Moron R, Zarzuelo A, and Galisteo M. Long-term
2: 23–27, 2007.
resveratrol administration reduces metabolic disturbances
133. Soares AF, Guichardant M, Cozzone D, Bernoud-Hubac N,
and lowers blood pressure in obese Zucker rats. Biochem
Bouzaidi-Tiali N, Lagarde M, and Geloen A. Effects of
Pharmacol 77: 1053–1063, 2009.
oxidative stress on adiponectin secretion and lactate pro-
119. Ruige JB, Assendelft WJ, Dekker JM, Kostense PJ, Heine RJ,
duction in 3T3-L1 adipocytes. Free Radic Biol Med 38: 882–
and Bouter LM. Insulin and risk of cardiovascular disease:
a meta-analysis. Circulation 97: 996–1001, 1998.
134. Staiger H and Haring HU. Adipocytokines: fat-derived
120. Saito Y, Yokoyama M, Origasa H, Matsuzaki M, Matsu-
humoral mediators of metabolic homeostasis. Exp Clin
zawa Y, Ishikawa Y, Oikawa S, Sasaki J, Hishida H, Itakura
Endocrinol Diabetes 113: 67–79, 2005.
H, Kita T, Kitabatake A, Nakaya N, Sakata T, Shimada K,
135. Stauss HM, Godecke A, Mrowka R, Schrader J, and Persson
and Shirato K. Effects of EPA on coronary artery disease in
PB. Enhanced blood pressure variability in eNOS knockout
hypercholesterolemic patients with multiple risk factors:
mice. Hypertension 33: 1359–1363, 1999.
sub-analysis of primary prevention cases from the Japan
136. Swindell WR. Comparative analysis of microarray data
EPA Lipid Intervention Study ( JELIS). Atherosclerosis 200:
identifies common responses to caloric restriction among
135–140, 2008.
mouse tissues. Mech Ageing Dev 129: 138–153, 2008.
121. Sakurai T, Izawa T, Kizaki T, Ogasawara JE, Shirato K,
137. Tabas I. Apoptosis and efferocytosis in mouse models of
Imaizumi K, Takahashi K, Ishida H, and Ohno H. Exercise
atherosclerosis. Curr Drug Targets 8: 1288–1296, 2007.
training decreases expression of inflammation-related adi-
138. Tamori Y, Sakaue H, and Kasuga M. RBP4, an unexpected
pokines through reduction of oxidative stress in rat white
adipokine. Nat Med 12: 30–31; discussion 31, 2006.
adipose tissue. Biochem Biophys Res Commun 379: 605–609,
139. Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, Lopez
BL, Koch W, Chan L, Goldstein BJ, and Ma XL. Adipo-
122. Sarzani R, Salvi F, Dessi-Fulgheri P, and Rappelli A. Renin-
angiotensin system, natriuretic peptides, obesity, metabolic
reperfusion involves the reduction of oxidative/nitrative
syndrome, and hypertension: an integrated view in hu-
stress. Circulation 115: 1408–1416, 2007.
mans. J Hypertens 26: 831–843, 2008.
140. Tarpey MM. Sepiapterin treatment in atherosclerosis. Ar-
123. Sasaki H, Asanuma H, Fujita M, Takahama H, Wakeno M,
terioscler Thromb Vasc Biol 22: 1519–1521, 2002.
Ito S, Ogai A, Asakura M, Kim J, Minamino T, Takashima
141. Thomas SR, Witting PK, and Drummond GR. Redox con-
S, Sanada S, Sugimachi M, Komamura K, Mochizuki N,
trol of endothelial function and dysfunction: molecular
and Kitakaze M. Metformin prevents progression of heart
mechanisms and therapeutic opportunities. Antioxid Redox
failure in dogs: role of AMP-activated protein kinase. Cir-
Signal 10: 1713–1765, 2008.
culation 119: 2568–2577, 2009.
142. Thony B, Auerbach G, and Blau N. Tetrahydrobiopterin
124. Scheen AJ. Prevention of type 2 diabetes mellitus through
biosynthesis, regeneration and functions. Biochem J 347 Pt 1:
inhibition of the Renin-Angiotensin system. Drugs 64:
1–16, 2000.
2537–2565, 2004.
143. Thorne RF, Mhaidat NM, Ralston KJ, and Burns GF. CD36
125. Schulz E, Jansen T, Wenzel P, Daiber A, and Munzel T.
is a receptor for oxidized high density lipoprotein: impli-
Nitric oxide, tetrahydrobiopterin, oxidative stress, and en-
cations for the development of atherosclerosis. FEBS Lett
dothelial dysfunction in hypertension. Antioxid Redox Signal
581: 1227–1232, 2007.
10: 1115–1126, 2008.
144. Thorp E and Tabas I. Mechanisms and consequences of
126. Sharma AM and Staels B. Review: peroxisome proliferator-
efferocytosis in advanced atherosclerosis. J Leukoc Biol 86:
activated receptor gamma and adipose tissue—understanding
1089–1095, 2009.
obesity-related changes in regulation of lipid and glucose
144a.UK Prospective Diabetes Study (UKPDS) Group. Effect of
metabolism. J Clin Endocrinol Metab 92: 386–395, 2007.
intensive blood-glucose control with metformin on com-
OXIDATIVE STRESS AND METABOLIC SYNDROME
plications in overweight patients with type 2 diabetes
drome in obese individuals: the impact of rapid weight loss
(UKPDS 34). Lancet 352: 854–865, 1998.
through caloric restriction. J Clin Endocrinol Metab 89: 2697–
145. Vanhoutte PM. Endothelial dysfunction: the first step to-
ward coronary arteriosclerosis. Circ J 73: 595–601, 2009.
159. Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida
146. Vasquez-Vivar J, Duquaine D, Whitsett J, Kalyanaraman B,
S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y,
and Rajagopalan S. Altered tetrahydrobiopterin metabo-
Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R,
lism in atherosclerosis: implications for use of oxidized
Kahn BB, and Kadowaki T. Adiponectin stimulates glucose
tetrahydrobiopterin analogues and thiol antioxidants. Ar-
utilization and fatty-acid oxidation by activating AMP-
terioscler Thromb Vasc Biol 22: 1655–1661, 2002.
activated protein kinase. Nat Med 8: 1288–1295, 2002.
147. Vasquez-Vivar J, Martasek P, Whitsett J, Joseph J, and
160. Yassine HN, Marchetti CM, Krishnan RK, Vrobel TR,
Kalyanaraman B. The ratio between tetrahydrobiopterin
Gonzalez F, and Kirwan JP. Effects of exercise and caloric
and oxidized tetrahydrobiopterin analogues controls su-
restriction on insulin resistance and cardiometabolic risk
peroxide release from endothelial nitric oxide synthase: an
factors in older obese adults—a randomized clinical trial. J
EPR spin trapping study. Biochem J 362: 733–739, 2002.
Gerontol A Biol Sci Med Sci 64: 90–95, 2009.
148. Vieira VJ, Valentine RJ, Wilund KR, Antao N, Baynard T,
161. Yokoyama M, Origasa H, Matsuzaki M, Matsuzawa Y,
and Woods JA. Effects of exercise and low-fat diet on adi-
Saito Y, Ishikawa Y, Oikawa S, Sasaki J, Hishida H, Itakura
pose tissue inflammation and metabolic complications in
H, Kita T, Kitabatake A, Nakaya N, Sakata T, Shimada K,
obese mice. Am J Physiol Endocrinol Metab 296: E1164–E1171,
and Shirato K. Effects of eicosapentaenoic acid on major
coronary events in hypercholesterolaemic patients ( JELIS):
149. Vinayagamoorthi R, Bobby Z, and Sridhar MG. Anti-
a randomised open-label, blinded endpoint analysis. Lancet
oxidants preserve redox balance and inhibit c-Jun-N-
369: 1090–1098, 2007.
terminal kinase pathway while improving insulin signaling
162. You T and Nicklas BJ. Effects of exercise on adipokines and
in fat-fed rats: evidence for the role of oxidative stress on
the metabolic syndrome. Curr Diab Rep 8: 7–11, 2008.
IRS-1 serine phosphorylation and insulin resistance. J En-
163. Zafon C. Fat and aging: a tale of two tissues. Curr Aging Sci
docrinol 197: 287–296, 2008.
2: 83–94, 2009.
150. Viollet B, Guigas B, Leclerc J, Hebrard S, Lantier L, Mounier
164. Zhang H, Zhang J, Ungvari Z, and Zhang C. Resveratrol
R, Andreelli F, and Foretz M. AMP-activated protein kinase
improves endothelial function: role of TNF{alpha} and
in the regulation of hepatic energy metabolism: from
vascular oxidative stress. Arterioscler Thromb Vasc Biol 29:
physiology to therapeutic perspectives. Acta Physiol (Oxf)
1164–1171, 2009.
196: 81–98, 2009.
165. Zhou Q and Liao JK. Rho kinase: an important mediator of
151. Wainwright CL. Matrix metalloproteinases, oxidative stress
atherosclerosis and vascular disease. Curr Pharm Des 15:
and the acute response to acute myocardial ischaemia and
3108–3115, 2009.
reperfusion. Curr Opin Pharmacol 4: 132–138, 2004.
166. Zhu M, Lee GD, Ding L, Hu J, Qiu G, de Cabo R, Bernier M,
152. Waki H and Tontonoz P. Endocrine functions of adipose
Ingram DK, and Zou S. Adipogenic signaling in rat white
tissue. Annu Rev Pathol 2: 31–56, 2007.
adipose tissue: modulation by aging and calorie restriction.
153. Wang G, Woo CW, Sung FL, Siow YL, and O K. Increased
Exp Gerontol 42: 733–744, 2007.
monocyte adhesion to aortic endothelium in rats with hy-
167. Zhu M, Miura J, Lu LX, Bernier M, DeCabo R, Lane MA,
perhomocysteinemia: role of chemokine and adhesion
Roth GS, and Ingram DK. Circulating adiponectin levels
molecules. Arterioscler Thromb Vasc Biol 22: 1777–1783, 2002.
increase in rats on caloric restriction: the potential for in-
154. Wang Y, Gao E, Tao L, Lau WB, Yuan Y, Goldstein BJ,
sulin sensitization. Exp Gerontol 39: 1049–1059, 2004.
Lopez BL, Christopher TA, Tian R, Koch W, and Ma XL.
168. Ziemke F and Mantzoros CS. Adiponectin in insulin re-
AMP-activated protein kinase deficiency enhances myo-
sistance: lessons from translational research. Am J Clin Nutr
cardial ischemia/reperfusion injury but has minimal effect
91: 258S–261S.
on the antioxidant/antinitrative protection of adiponectin.
Circulation 119: 835–844, 2009.
155. Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar
M, and Sinclair D. Sirtuin activators mimic caloric restric-
Address correspondence to:
tion and delay ageing in metazoans. Nature 430: 686–689,
Second Department of Internal Medicine
156. Worz CR and Bottorff M. Treating dyslipidemic patients
Kansai Medical University
with lipid-modifying and combination therapies. Pharma-
cotherapy 23: 625–637, 2003.
Moriguchi City 570-8507
157. Wu HC, Chen SY, Shroff SG, and Carroll JD. Stress analysis
using anatomically realistic coronary tree. Med Phys 30:2927–2936, 2003.
158. Xydakis AM, Case CC, Jones PH, Hoogeveen RC, Liu MY,
Smith EO, Nelson KW, and Ballantyne CM. Adiponectin,
Date of first submission to ARS Central, November 4, 2010;
inflammation, and the expression of the metabolic syn-
date of acceptance, December 2, 2010.
Abbreviations Used
iNOS ¼ inducible nitric oxide synthase
ACE ¼ angiotensin-converting enzyme
IRS ¼ insulin receptor substrate
ACEI ¼ angiotensin-converting inhibitor
LDL ¼ low-density lipoprotein
AMPK ¼ AMP-activated protein kinase
MCP-1 ¼ monocyte chemoattractant protein-1
Ang II ¼ angiotensin II
MetS ¼ metabolic syndrome
ARBs ¼ angiotensin II type-1 receptor blockers
NO ¼ nitric oxide
BH2 ¼ dihydrobiopterin
OxLDL ¼ oxidized LDL
BH4 ¼ tetrahydrobiopterin
PAI-1 ¼ plasminogen activator inhibitor-1
CR ¼ caloric restriction
PI3K ¼ phosphatidylinositol 3-kinase
DPP-4 ¼ dipeptidyl peptidase-IV
PPAR ¼ peroxisome proliferator activated receptor
eNOS ¼ endothelial nitric oxide synthase
RAAS ¼ renin-angiotensin aldosterone system
EPA ¼ eicosapentaenoic acid
ROCK ¼ Rho-kinase
GLUT4 ¼ glucose transporter-4
ROS ¼ reactive oxygen species
GTPCH ¼ guanosine triphosphate cyclohydrolase
SMCs ¼ smooth muscle cells
HDL ¼ high-density lipoprotein
T2DM ¼ type-2 diabetes mellitus
HTN ¼ hypertension
TDZs ¼ thiazolidinediones
IGT ¼ impaired glucose tolerance
TNF-a ¼ tumor necrosis factor-a
This article has been cited by:
1. Nilanjana Maulik , Juan A. Sanchez . 2011. Risk Factors in Heart Disease: Therapeutic Interventions. Antioxidants & Redox
Signaling 15:7, 1765-1767. [
Source: http://www.eurodream.net/files/Oxidative%20Stress%20as%20Pathogenesis%20of%20Cardiovascular%20Risk%20Associated%20with%20Metabolic%20Syndrome%20(2).pdf
[ Evidence-Based Medicine ] Antithrombotic Therapy for VTE DiseaseCHEST Guideline and Expert Panel Report Clive Kearon, MD, PhD; Elie A. Akl, MD, MPH, PhD; Joseph Ornelas, PhD; Allen Blaivas, DO, FCCP; David Jimenez, MD, PhD, FCCP; Henri Bounameaux, MD; Menno Huisman, MD, PhD; Christopher S. King, MD, FCCP; Timothy A. Morris, MD, FCCP; Namita Sood, MD, FCCP; Scott M. Stevens, MD; Janine R. E. Vintch, MD, FCCP; Philip Wells, MD; Scott C. Woller, MD;
Agroforest Syst (2007) 71:185–193DOI 10.1007/s10457-007-9071-8 A review of suitable companion crops for black walnut Robert Scott Æ William C. Sullivan Received: 9 May 2006 / Accepted: 16 May 2007 / Published online: 21 June 2007 ! Springer Science+Business Media B.V. 2007 Black walnut (Juglans nigra L.) is a temperate tree grown for nuts and wood, but it isallelopathic to certain plants and animals. We com-